
Special Article: Vibriosis
Austin J Vet Sci & Anim Husb. 2023; 10(2): 1118.
High Concordance between the Whole Genome Sequencing data Analysis Methods for Vibrio Cholerae Surveillance in Finland and Norway
Nyholm O1*, Tønnessen R2,4, Antony-Samy JK2, Halkilahti J1, Amato E3 and Salmenlinna S1
1Department of Health Security, Finnish Institute for Health and Welfare (THL), Finland
2Department of Infection Control and Vaccines, The Norwegian Institute of Public Health, Norway
3Department of Infection Control and Preparedness, The Norwegian Institute of Public Health, Norway
4European Public Health Microbiology Training Program (EUPHEM), European Centre for Disease Prevention and Control (ECDC), Sweden
*Corresponding author: Nyholm ODepartment of Health Security, Finnish Institute for Health and Welfare (THL), P.O Box 30, 00271 Helsinki, Finland
Received: January 23, 2023; Accepted: February 20, 2023; Published: February 27, 2023
Abstract
Vibrio cholerae infections, both vibriosis and cholera, are rare in Northern Europe. However, the coastal areas suitable for transmission have increased during recent years. Accessible and validated molecular diagnostic methods are needed to monitor such infections. Here, we describe the comparison and validation of Whole Genome Sequencing (WGS) data analysis methods for V. cholerae characterization in the public health institutes of Finland and Norway. The results showed a concordance of 96.7% between the methods in the two countries.
Keywords: Vibrio cholerae; Cholera; Vibriosis; Whole genome sequencing; Bioinformatics
Introduction
Cholera causes around 100,000 deaths each year globally [1]. In Northern Europe, classical cholera caused by toxin-producing Vibrio cholerae serotypes O1 and O139 is rare and almost exclusively travel-related. The occurrence of non-toxigenic, non-O1/non-O139 V. cholerae causing vibriosis varies and is more frequent during warm summers [2-4]. The coastal areas suitable for V. cholerae transmission increased substantially across countries between 2003 and 2019 due to climate change [5]. Therefore, molecular diagnostic methods able to detect and characterize V. cholera isolates are needed for laboratory preparedness purposes and for the surveillance of vibriosis and cholera.
We compared the Whole Genome Sequencing (WGS) data analysis methods for V. cholerae surveillance at the Finnish Institute for Health and Welfare (THL) and the Norwegian Institute of Public Health (NIPH). The methods consisted of species confirmation and virulence genes detection for V. cholerae in both countries with different bioinformatics software. Screening for six markers of toxigenic V. cholerae [6] were included. These markers were genes for detection of V. cholerae species (toxR), cholera toxin (ctxA), serogroups O1 (wbeO1) and O139 (wbfO139), and biotypes classical and El Tor (tcpA variants). The aim of the study was to validate the WGS data analysis methods at THL, Finland, using the pipeline that has previously been established at NIPH, Norway.
Materials and Methods
We included Vibrio spp. sequences from 392 isolates in the comparison of the WGS data analysis methods (Table 1). These sequences included publicly available sequences from the NCBI database, clinical and environmental isolates from Finland, and three reference isolates obtained from the culture collection of University of Gothenburg. The Finnish isolates and the reference isolates were sequenced at THL. Briefly, the DNA extraction was performed using Mag Attract kit (Qiagen), library preparation using Nextera XT kit (Illumina), and sequencing using Illumina MiSeq NGS platform with 300 cycles kit (Illumina)To assess the performance of THL’s WGS data analysis methods, the same sequences (fastq files) were run at NIPH using the existing bioinformatics pipeline [7]. The bioinformatics software algorithms used to analyze the WGS data in THL and NIPH have been previously published and they are described in (Figure 1). THL’s bioinformatics pipeline uses Kraken2 [8] for species confirmation and contamination check and ReMatch [9] to screen for the six V. cholerae specific marker genes toxR, cholera toxin ctxA, serogroups O1 (wbeO1) and O139 (wbfO139), and biotypes classical and El Tor (tcpA variants). The NCBI accession numbers for the target genes and their GC contents were as follows: toxR KF498634.1 GC 45.2%, ctxA AF463401.1 GC 38.4%, wbeO1 KC152957.1 GC 39.6%, wbfO139 AB012956.1 GC 42.0%, tcpA_Classical M33514.1GC 40.3%, and tcpA_El_Tor KP187623.1 GC 43.4%. NIPH’s pipeline also used Kraken2 for species confirmation and contamination check, while ARIBA [10] was used for mapping the reads to databases and it was supplemented with a Python module to screen for the six V. cholerae marker genes. For ARIBA, the databases were custom made using the fasta sequences of the six marker genes. ARIBA uses Bowtie2 to map the reads to databases. ReMatch maps reads, utilizing also Bowtie2, onto a set of reference sequences to determine if the chosen loci of interest are either absent or present in a sample. The outcome was determined by evaluating sequencing depth as well as coverage and similarity when compared to the original reference sequence. The outputs from both pipelines were analyzed using a threshold of ≥80% coverage for the six markers.The results and the interpretation obtained using the methods at NIPH were compared with those at THL.
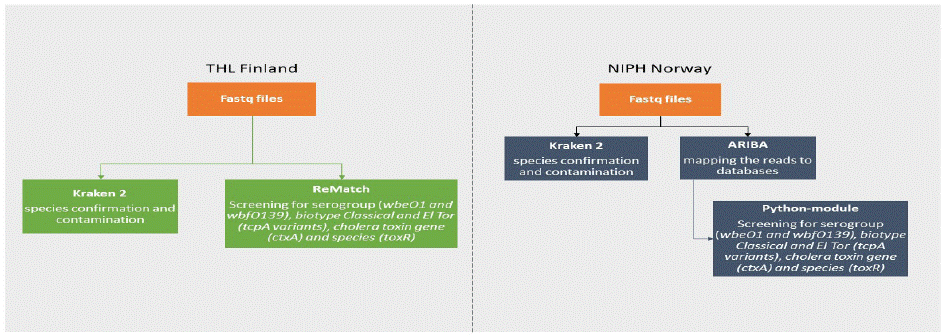
Figure 1: Whole genome sequencing pipelines for identification and typing of Vibrio cholerae at the public health institutes of Finland (THL) and Norway (NIPH).
The sequence reads’ quality was assessed at NIPH and THL using FastQC/MultiQC [11]. Regardless of the quality, all the sequences were included in the study.
Results and Discussion
Out of the 392 sequences, 379 had an identical typing result for the identified Vibrio species with Kraken2 and for the six V. cholerae marker genes both in THL and NIPH reaching an overall concordance of 96.7% (Supplementary Table 1). Only 13 sequences had a discordant typing result (Table 2).
![]()
NCBI
Study accession PRJEB14630:
ERR1485291 ERR1485293 ERR1485295 ERR1485297Study accession PRJEB303115:
SRR3020407SRR3020408SRR3020409 SRR3020410 SRR3020411 SRR3020412 SRR3020413 SRR3020414 SRR3020415 SRR3020416 SRR3020417 SRR3020418 SRR3020419 SRR3020420 SRR3020421 SRR3020422 SRR3020423 SRR3020424 SRR3020425 SRR3020426 SRR3020427 SRR3020428 SRR3020429 SRR3020430 SRR3020431 SRR3020432 SRR3020433 SRR3020434 SRR3020435 SRR3020436 SRR3020437 SRR3020438 SRR3020439 SRR3020440 SRR3020441 SRR3020442 SRR3020443 SRR3020444 SRR3020445 SRR3020446 SRR3020447 SRR3020448 SRR3020449 SRR3020450 SRR3020451 SRR3020452 SRR3020453 SRR3020454 SRR3020455 SRR3020456 SRR3020457 SRR3020458 SRR3020459 SRR3020460 SRR3020461 SRR3020462 SRR3020463SRR3020464 SRR3020465 SRR3020466 SRR3020467 SRR3020468 SRR3020469 SRR3020470 SRR3020471 SRR3020472 SRR3020473 SRR3020474 SRR3020475 SRR3020476 SRR3020477 SRR3020478 SRR3020479SRR3020480 SRR3020481 SRR3020482 SRR3020483 SRR3020484 SRR3020485 SRR3020486 SRR3020487 SRR3020488 SRR3020489 SRR3020490 SRR3020491 SRR3020492 SRR3020493 SRR3020494 SRR3020495 SRR3020496 SRR3020497 SRR3020498 SRR3020499 SRR3020500 SRR3020501 SRR3020502 SRR3020503 SRR3020504 SRR3020505 SRR3020506 SRR3020507 SRR3020508 SRR3020509 SRR3020510 SRR3020511 SRR3020512 SRR3020513 SRR3020514SRR3020515 SRR3020516 SRR3020517 SRR3020518 SRR3020519 SRR3020520 SRR3020521 SRR3020522 SRR3020523 SRR3020524SRR3020525 SRR3020526 SRR3020527 SRR3020528 SRR3020529 SRR3020530 SRR3020531 SRR3020532 SRR3020533 SRR3020534 SRR3020535 SRR3020536 SRR3020537 SRR3020538 SRR3020539 SRR3020540 SRR3020541 SRR3020542 SRR3020543 SRR3020544 SRR3020545SRR3020546 SRR3020547 SRR3020548SRR3020549 SRR3020550 SRR3020551 SRR3020552 SRR3020553 SRR3020554 SRR3020555 SRR3020556 SRR3020557 SRR3020558 SRR3020559 SRR3020560 SRR3020562 SRR3020563 SRR3020564 SRR3020565 SRR3020566 SRR3020567 SRR3020568 SRR6027644 SRR6027645 SRR6027646 SRR6027647 SRR6027649 SRR6027655 SRR6027656 SRR6027663 SRR6027664 SRR6027665 SRR6027688 SRR6027689 SRR6027690 SRR6027691 SRR6027692 SRR6027693 SRR6027694 SRR6027695 SRR6027696 SRR6027697 SRR7062492 SRR7062493 SRR7062494 SRR7062495 SRR7062496 SRR7062497 SRR7062498 SRR7062499 SRR7062500 SRR7062501 SRR7062502 SRR7062503 SRR7062504 SRR7062505 SRR7062506 SRR7062507 SRR7062508 SRR7062509 SRR7062510 SRR7062511 SRR7062512 SRR7062513 SRR7062514 SRR7062515 SRR7062516 SRR7062517 SRR7062518 SRR7062519 SRR7062520 SRR7062521 SRR7062522 SRR7062523 SRR7062524 SRR7062525 SRR7062526 SRR7062527 SRR7062528 SRR7062529 SRR7062530 SRR7062531 SRR7062532 SRR7062533 SRR7062534 SRR7062535 SRR7062536 SRR7062537 SRR7062538 SRR7062539 SRR7062540 SRR7062541 SRR7062542 SRR7062543 SRR7062544 SRR7062545 SRR7062546 SRR7062547 SRR7062548 SRR7062549 SRR7062550 SRR7062551 SRR7062552 SRR7062553 SRR7062554 SRR7062555 SRR7062556 SRR7062557 SRR7062558 SRR7062559 SRR7062560 SRR7062561 SRR7062562 SRR7062563 SRR7062564 SRR7062565 SRR7062566 SRR7062567 SRR7062568 SRR7062569 SRR7062570 SRR7062571 SRR7062572 SRR7062573 SRR7062574 SRR7062575 SRR7062576 SRR7062577 SRR7062578SRR7062579 SRR7062580 SRR7062581 SRR7062582 SRR7062583 SRR7062584 SRR7062585 SRR7062586 SRR7062587 SRR7062588 SRR7062589 SRR7062590 SRR7062591 SRR7062592 SRR7062593 SRR7062594 SRR7062595 SRR7062596 SRR7062597 SRR7062598 SRR7062599 SRR7062600 SRR7062601 SRR7062602 SRR7062603 SRR7062604 SRR7062605 SRR7062606 SRR7062607 SRR7062608 SRR7062609 SRR7062610 SRR7062611 SRR7062612 SRR7062613 SRR7062614 SRR7062615 SRR7062616 SRR7062617 SRR7062618 SRR7062619 SRR7062620 SRR7062621 SRR7062622 SRR7062623 SRR7062624 SRR7062625 SRR7062626 SRR7062627 SRR7062628 SRR7062629 SRR7062630 SRR7062631 SRR7062632 SRR7062633 SRR7062634 SRR7062635 SRR7062636 SRR7062637 SRR7062638 SRR7062639 SRR7062640 SRR7062641 SRR7062642 SRR7062643 SRR7791638 SRR8668523 SRR8668524THL
Study accession PRJEB52536:
P03-D06-125831 P03-G03-125196 P03-F05-125602 P03-G01-122970 P03-D05-125359 P03-D02-125109 P03-E02-125118 P03-H03-125197 P03-G06-125834 P03-F03-125174 P03-H04-125251 P03-F01-118295 P03-B01-84440 P03-G02-125155 P03-E04-125222 P03-F04-125223 P03-A03-125157 P03-A02-125098 P03-D01-104751 P03-B02-125099 P03-C04-125217 P03-B03-125161 P03-E03-125173 P03-H02-125156 P03-B04-125205 P03-A04-125204 P03-G05-125826 P03-C01-84456 P03-E05-125481 P03-E01-118275 P03-C03-125168 P03-E06-125832 P03-F06-125833 P03-H06-125843 P03-C06-125830 P03-A01-63347 P03-A07-125844 P03-H05-125827 P03-D04-125218 P03-C02-125108 P03-A05-125276 P03-B05-125277 P03-C05-125305 P03-D03-125172 P03-H01-124364 P03-F02-125151 P03-G04-125224 P03-A06-125828 P03-B06-125829*Contact information: CCUG; CULTURE COLLECTION UNIVERSITY OF GÖTEBORG, Department of Clinical Bacteriology, Guldhedsgatan 10, SE-413 46 Göteborg, Tel +46.31-342 47 02, Fax:+46.31-82 96 17, ccug@ccug.se, www.ccug.se
Table 1: Accession numbers for the 392 sequences included in the study obtained from NCBI (n=340), Finnish isolates sequenced at THL (n=49, of which 32 were clinical V. cholerae isolates from the year 2016 to 2018 and 13 environmental V. cholerae isolates, and four were other vibrios (V. alginolyticus, Vibrio ordalii/Vibrio anguillarum, V. parahaemolyticus, V. vulnificus) and toxigenic V. cholerae reference strains (n=3) obtained from Culture Collection from the University of Gothenburg (CCUG) and sequenced at THL. The source of isolation for the reference strains and for the sequences obtained from NCBI is unknown.
The following results, using three different sources, were obtained: (i) a concordance of 96.2% for the 340 isolate sequences from the public databases as 327 out of 340 analysed sequences had an identical typing result; (ii) a concordance of 100% for the 49 isolates from THL’s culture collection; (iii) a concordance of 100% for the three isolates obtained by THL from the culture collection of University of Gothenburg.
The 13 discordant results were all among the sequences derived from public databases. The species identification of the V. cholerae- specific toxR marker was not always concordant between THL and NIPH. Further, there seemed to be discordant results among the serogroup O139, ctxA-toxin, and the biotype marker gene of El Tor. At least some of these discrepancies could be linked to the quality of the sequences and to the definition of the threshold of ≥80% coverage for the marker genes used for the analysis.A closer examination of the results indicated low quality and/or low coverage (less than 30x) for some of these 13 sequences. For most of the sequences that were interpreted as non-V. cholerae either at THL or NIPH, the percentage of V. cholerae assigned reads was low (less than 70%) in Kraken2. We cannot guarantee the quality of the sequences deposited in the public databases even though some of them have been previously described and used in the validation of a WGS-based typing method [6].In addition, some of the discrepancies may be due to using different algorithms and the methods the pipelines are dependent on at THL and NIPH, mainly ReMatch and ARIBA, respectively. It should also be noted that Illumina’s library preparation kit Nextera XT was used to sequence most of the 13 sequences with discordant results (Table 2), which is known to result in uneven genome-wide sequencing coverage and poor performance on low and high GC-content regions compared to Illumina’s newer DNA Prep Kit [12].
![]()
Sample ID
Institute
toxR
ctxA
wbeO1
El Tor
Classical
wbeO139
Kraken 2 % of V. cholerae assigned reads
Contaminationcheck
Coverage
Averagereadlenght
No of reads
Library preparation kit (information obtained from NCBI)
Qualitycheckcomments
SRR3020479
NIPH
Vibrio cholerae 96,76
toxigenic 99,49
non-O1
El Tor 99,26
non-Classical
O139 99,93
83,13
no contamination
0.0908457
88.1884
4155
Notdeclared
Very low coverage, very low number of reads
THL
Non-Vibrio cholerae 0,0
non-toxigenic 0,0
non-O1
non-El Tor 0,0
non-Classical
non-O139 0,0
SRR3020534
NIPH
Vibrio cholerae
toxigenic 96,27
non-O1
El Tor
non-Classical
O139
79,75
no contamination
49.0262
88.9863
2222201
Notdeclared
OK
THL
Vibrio cholerae
non-toxigenic 0,0
non-O1
El Tor
non-Classical
O139
SRR3020560
NIPH
Non-Vibrio cholerae 39,62
toxigenic
non-O1
non-El Tor 67,70
non-Classical
non-O139 73,56
83,83
no contamination
24.3535
88.5201
1109678
Notdeclared
Low genome coverage
THL
Vibrio cholerae 99,1
toxigenic
non-O1
El Tor 100,0
non-Classical
O139 100,0
SRR3020562
NIPH
Non-Vibrio cholerae 24,13
non-toxigenic 68,98
non-O1
non-El Tor 34,81
non-Classical
non-O139 64,32
81,64
no contamination
4.22518
88.2902
193024
Notdeclared
Very low coverage, low number of reads
THL
Vibrio cholerae 91,92
toxigenic 100,0
non-O1
El Tor 91,7
non-Classical
O139 92,21
SRR7062498
NIPH
Non-Vibrio cholerae 47,59
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
76,09
no contamination
39.9582
96.9775
1661931
Nextera XT
OK
THL
Vibrio cholerae 83,61
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
SRR7062528
NIPH
Non-Vibrio cholerae 36,36
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
66,25
no contamination
20.1762
99.2608
819860
Nextera XT
Low coverage
THL
Vibrio cholerae 82,60
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
SRR7062552
NIPH
Non-Vibrio cholerae 43,10
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
64,8
no contamination
31.3243
97.9996
1289247
Nextera XT
OK
THL
Vibrio cholerae 80,13
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
SRR7062584
NIPH
Non-Vibrio cholerae 43,43
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
62,29
no contamination
53.546
97.7773
2208855
Nextera XT
OK
THL
Vibrio cholerae 82,83
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
SRR7062585
NIPH
Vibrio cholerae
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139 77,64
56,66
no contamination
57.4894
100.006
2318676
Nextera XT
OK
THL
Vibriocholerae
non-toxigenic
non-O1
non-El Tor
non-Classical
O139 82,36
SRR7062596
NIPH
Non-Vibrio cholerae 41,75
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
74,76
no contamination
52.5005
98.854
2142137
Nextera XT
OK
THL
Vibrio cholerae 82,94
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
SRR7062609
NIPH
Non-Vibrio cholerae 41,08
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
76,17
no contamination
33.7214
99.5241
1366642
Nextera XT
OK
THL
Vibrio cholerae 82,83
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
SRR7062622
NIPH
Non-Vibrio cholerae 35,47
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
63,48
no contamination
42.963
99.9761
1733309
Nextera XT
OK
THL
Vibrio cholerae 83,61
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
SRR7062643
NIPH
Non-Vibrio cholerae 75,65
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
69,64
no contamination
6.23164
95.0902
264329
Nextera XT
Very low coverage, low number of reads
THL
Vibrio cholerae 83,73
non-toxigenic
non-O1
non-El Tor
non-Classical
non-O139
Table 2: Summary of the 13 V. Cholerae sequences with discordant results for some of the marker genes (in grey) analysed with the methods used at NIPH and THL. The percentage of the target gene coverage is reported in case of discordant results.
The study material consisted of a limited number of laboratory-confirmed toxigenic V. cholerae isolates from Finland since Vibrio infections are rare. Therefore, we supplemented the dataset with three toxigenic reference isolates supplied from the culture collection of University of Gothenburg. Another limitation of the study concerns non-V. cholerae species confirmation. Only four laboratory-confirmed isolates from THL’s culture collection were available for the WGS data analysis. Thus, we cannot conclude that Kraken2 can verify the non-V. cholerae species accurately. Despite these limitations, the results of this study show a clear advantage of collaboration between national public health institutes as reaching a more harmonized molecular approach for Vibrio characterization between Finland and Norway.
Conclusion
The high concordance between the WGS-based analysis methods highlights that the selected tools in the two public health institutes are suitable for typing of V. cholerae. The selected method for WGS-based typing of V. cholerae was validated to be used at THL’s reference laboratory for national V. cholerae surveillance. Limitations of the WGS-based method were observed in the detection of marker genes with low-quality sequences. Therefore, it is essential to monitor the quality of sequencing to achieve reliable results regardless of analysis methods and site.
Acknowledgements
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. We would like to acknowledge Aftab Jasir (ECDC), Umaer Naseer (NIPH) and Astrid Louise Wester (NIPH) for providing their valuable insight and comments.
References
- Clemens JD, Nair GB, Ahmed T, Qadri F, Holmgren J. Cholera. Lancet. 2017; 390: 1539-1549.
- Andersson Y, Ekdahl K. Wound infections due to Vibrio cholerae in Sweden after swimming in the Baltic Sea, summer 2006. Euro Surveill. 2006; 11: E060803.2.
- Baker-Austin C, Trinanes JA, Salmenlinna S, Löfdahl M, Siitonen A, et al. Heat wave-associated vibriosis, Sweden and Finland, 2014. Emerging Infectious Diseases. 2016; 22: 1216-1220.
- Amato E, Riess M, Thomas-Lopez D, Linkevicius M, Pitkänen T, et al. Epidemiological and microbiological investigation of the large increase of vibriosis in northern Europe in 2018. Eurosurveillance. 2022; 27: 2101088.
- Romanello M, McGushin A, Di Napoli C, Drummond P, Hughes N, et al. The 2021 report of the Lancet Countdown on health and climate change: code red for a healthy future. Lancet. 2021; 398: 1619-1662.
- Greig DR, Schaefer U, Octavia S, Hunter E, Chattaway MA, Dallman TJ, et al. Evaluation of whole-genome sequencing for identification and typing of Vibrio cholerae. Journal of Clinical Microbiology. 2018; 56: e00831-18.
- The Norwegian Institute of Public Health. Vibrio project. GitHub.
- Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biology. 2019; 20: 257.
- Machado MP, Ribeiro-Goncalves B, SilvaM, MendesI, RossiM, RamirezM, et al. ReMatCh. GitHub.
- Hunt M, Mather AE, Sánchez-Busó L, Page AJ, Parkhill J, Keane JA, et al. ARIBA: rapid antimicrobial resistance genotyping directly from sequencing reads. Microbial Genomics. 2017; 3: e000131.
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010.
- Poates A, Truong J, Lindsey R, Griswold T, Williams-Newkirk AJ, et al. Sequencing of Enteric Bacteria: Library Preparation Procedure Matters for Accurate Identification and Characterization. Foodborne pathogens and Disease. 2022; 19:569-578.