
Review Article
Austin J Vet Sci & Anim Husb. 2024; 11(2): 1143.
Reverse Vaccinology Approach Against Viruses: A Review
Adefris Zeniye¹; Ibsa Teshome²*
Department of Veterinary laboratory technology, Ambo University, Ambo, Ethiopia
*Corresponding author: Adefris Zeniye Department of Veterinary laboratory technology, Ambo University, Ambo, Po Box: 19, Ethiopia. Tel: +251921368315; Fax: 2510112365639 Email: adefriszeniye2020@gmail.com
Received: February 14, 2024 Accepted: March 29, 2024 Published: april 05, 2024
Summary
Vaccine design is a complex process. However, progresses in bioinformatics will probably make vaccine design and drug development easy. Vaccine design using reverse vaccinology approach is a quick as well as inexpensive than conventional vaccinology. Therefore, the objective of this manuscript is to review the reverse vaccinology approach against viral vaccine development. Reverse vaccinology against viruses is based on the information contained in the database found online from which protein sequences can be obtained without the need to grow the organism. Using this information, it is possible to select, identify, predict and analyze the target information according to the desire. That desirable information to construct vaccine is epitopes that are the immunologically active region of viruses. Since new, emerging diseases need fast measurement, reverse vaccinology approach utilizes bioinformatic tools like VaxiJen2.0 (which is able to calculate antigenic proteins), major histocompatibility complex class I and II prediction tools that helps to predict epitopes from the information contained in the protein data bank found online without the need to cultivate the virus which is a tiresome process. Therefore, based on this concept to construct vaccine against viruses reverse vaccinology approach should be followed.
Keywords: Bioinformatics tools; Conventional Vaccinology; Database; Epitopes; Reverse Vaccinology
Abbreviations: RV: Reverse Vaccinology; WHO: World Health Organization; MERS-COV: Middle East Respiratory Syndrome Corona Virus; FASTA: Fast Adaptive Shrinkage Threshold Algorithm; MEV: Multiepitope based Vaccine; RSV: Respiratory Syncytial Virus; SARS-COV: Severe Acute Respiratory Syndrome Corona Virus; HCOV: Human Corona Virus; MHC: Major Histocompatibility Complex; IEDB: Immune Epitope Data Base; DENV: Dengue Virus; HPV: Human Papilloma Virus; NCBI: National Center for Biotechnology Information; PyMOL: Python languge Molecule; RasMOL: Raster of a Molecule; PSIPRED: PSI-blast based Secondary Structure Prediction; PEP-FOLD: Peptide Fold; HLA: Human Leukocytes Antigen; PDBQT: Protein Data Bank, partial charge (Q), & Atom Type (T); PDB: Protein data bank; TLR4: Toll-like Receptor 4; CTL: Cytotoxic T Lymphocyte; HTL: Helper T Lymphocyte; BCL: B cell Lymphocyte; PADRE: Pedigree-Aware Distant-Relationship Estimation.
Introduction
Vaccines have transformed public health, particularly since national programmes for immunization first became properly established and coordinated in the 1960s [1]. A vaccine is a biological product that can be used to safely induce an immune response that confers protection against infection and/or disease on subsequent exposure to a pathogen [2]. Vaccines exploit the extraordinary ability of the highly evolved human immune system to respond to, and remember, encounters with pathogen antigens. However, for much of history, vaccines have been developed through empirical research without the involvement of immunologists [3]. Vaccine design is a complex process; however, progresses in bioinformatics will probably make vaccine design and drug development easy [4]. The design of vaccines can be divided into two broad categories: the traditional and the modern approach [5]. Traditional methods may take decades to unravel pathogens and antigens, diseases and immunity. However, modern approaches can be very fast, allowing identifying new vaccines for testing in only a few years [6]. Due to limitations of the conventional technology [7], modern technologies have come into existence, from which Reverse Vaccinology (RV) is one of the new technologies for vaccine development which represents a genome-based approach to vaccine development and developed for the first time in early 1990's by Rappuoli [8] to identify meningococcal protein vaccine candidates in Group B meningococcus (MenB) [9].
In reverse vaccinology, various tools of in silico biology are used to discover the novel antigens by studying the genetic makeup of a pathogen and the genes that could lead to good epitopes are determined. This method is a quick easy and cost-effective way to design vaccine [10]. It is a process of vaccine development where the novel antigens are identified by analyzing the genomic information of a virus or other organism [11]. RV offers two main advantages compared to traditional vaccine development approaches: identification of candidate antigens without the need to grow the pathogen and identification of any antigen independently by its purified quantity to be suitable for vaccine testing [12].
Therefore, the objective of this review is to highlight the reverse vaccinology approach against viral vaccine development.
Basic Concept of Vaccination
The history of vaccine production started with Edward Janner’s and Louis Pasteur’s innovation and immunization practices, making the development of vaccination a necessary practice for improving wealth [13]. The World Health Organization (WHO) has divided vaccines into three major categories: i) traditional, ii) innovator: new vaccines as RV; and iii) targeted. Traditional vaccines lead global market volume, and innovator vaccines drive global market value [14].
Vaccines, like natural infections, act by initiating an innate immune response, which in turn activates an antigen-specific adaptive immune response. Innate immunity is the first line of defence against pathogens that have entered the body. Adaptive immunity provides a second line of defence, generally at a later stage of infection, characterized by an extraordinarily diverse set of lymphocytes and antibodies able to recognize and eliminate virtually all known pathogens [15]. Vaccine design has made significant advances in the last century, evolving from serendipity to a more rational design due to advances in understanding immunological mechanisms and technology [16].
Vaccines can be produced using different processes. Vaccines may contain live attenuated pathogens (usually viruses), inactivated whole pathogens, toxoids (an inactivated form of the toxin produced by bacteria that causes the disease), or parts of the pathogens (e.g. natural or recombinant proteins, polysaccharides, conjugated polysaccharide or virus-like particles).
Reverse Vaccinology
Reverse Vaccinology refers to the concept of using genomic knowledge, without the actual cultivation of pathogen to determine the immunologically active components [17]. Pioneered by Dr. Rino Rappuoli, RV is an emerging vaccine development strategy that initiates a vaccine development from genome sequence bioinformatics analysis. It was first applied to development of a vaccine against serogroup B Neisseria meningitidis (MenB), the major cause of sepsis and meningitis in children and young adults [18].
Retrieval of Viral Protein Sequences
As suggested by [19], the generated genomic information is used to screen the inclusive set of potential proteins encoded by pathogens for the search of vaccine candidates. The availability of genomic information of the pathogen under study and, even the human or animal cell genome is an important pre-requirement for using RV. If the genome sequence is obtained, it is possible to identify all likely proteins that could be expressed [16,20].
During the study by [21] on Rational design of multi epitope-based subunit vaccine by exploring middle east respiratory syndrome corona virus (MERS-COV) proteome, Amino-acid sequence of structural (spike (AKL59401), membrane (AKL59407), Envelope (AKL59406) and nucleocapsid (AKL59408)) and non-structural proteins ( (A0A2P1ITC7) /1ab(A0A140AYZ4), NS3/3B/3C/3D/4A/4B/5, ORF3 (K9N796) /4a (K9N4V0) /4b (K9N643) /5(K9N7D2) /8 (A0A0U2GQ91)) [22,23] were obtained from Uniprot database in the fast adaptive shrinkage threshold algorithm (FASTA) format [24,25].
Epitope Selection
Epitopes are the immunologically active region of organisms which are recognized as foreign by the host responses and thus provide an excellent means for the production of an efficient vaccine because of their specificity towards a particular organism. An effective vaccine could be designed by the epitope prediction tools as the epitope stimulates immune reactions from both B cells and T cells (Kreiter et al., 2015). [26] suggested that epitopes selection is one of the critical steps for immunoinformatics study and during epitope selection we should select epitopes that are multi-specific and broad-based.
The research report by [27] summarized the general work flow with a set of immunoinformatics tools to design a Multiepitope Based Vaccine (MEV) against Respiratory Syncytial Virus (RSV) (Figure 1). A schematic representation of methodology and tools used in the present study for new vaccine development). Epitopes with the following characteristics are generally preferred to design a subunit vaccine: (a) highly antigenic, (b) immunogenic, (c) non-allergenic, (d) non-toxic, and (e) with significant population coverage [28- 30].
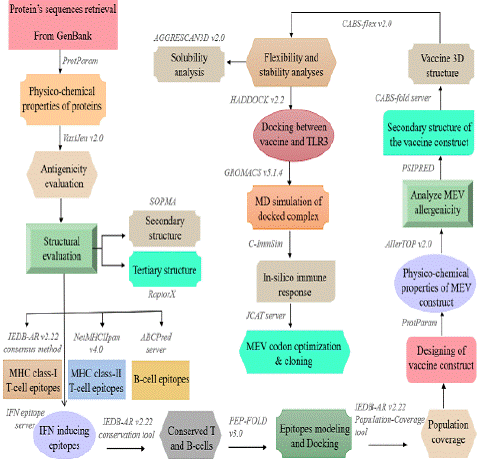
Figure 1: A schematic representation of methodology and tools used in the present study for new vaccine development [65].
Research done on viruses (Chikungunya and Mayaro) showed that to assess the best possible antigen, all the retrieved structural polyproteins of both of the viruses were subjected to computational antigenicity investigation in widely applied server known as VaxiJen2.0 (http://ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) which is able to calculate antigenic proteins more than 80% accurately [31]. The research report of [32] indicated the role of the Vaxign analysis framework [33,34] to compare the full proteomes of seven human coronavirus strains Severe acute respiratory syndrome 2 (SARS-CoV-2); SARS-CoV; MERS-CoV; Human coronavirus 229E (HCoV-229E); HCoV-OC43; HCoV-NL63 and HCoV-HKU1 [35].
Prediction of B-Cell and T-Cell Epitopes
The identification of immunodominant B- and T-cell epitopes that trigger protective immune responses in the host is critical for effective vaccine design [36,37]. The retrieved Homologous sequences of the antigenic proteins (i.e., capsid protein VP1 and protein VP2) of the Norovirus were used to perform multiple sequence alignment using Clustal Omega [38] and further for the prediction of T-cell epitope, Major Histocompatibility Complex I (MHC-I) (http://tools.iedb.org/mhci/) and MHC-II prediction tools (http: //tools. iedb.org/mhcii/) of immune epitope data base (IEDB) were used [39].
As a result, Epitopes that will bind with a higher number of MHC alleles are considered to be more immunogenic (Hajighahramani et al., 2017). As [40] confirmed, those epitopes that bind with the alleles involved in MERS-COV protection were chosen. B cell epitope prediction is concerned in finding the potential antigen that would interact with B lymphocytes and initiate an immune response [41]. As research done by [42] on vaccines against Dengue Virus 1 (DENV-1) and Human Papillomavirus 16 (HPV-16) indicated that the B cell lymphocytic epitopes were selected based on their lengths (the sequences with over ten amino acids in length were selected) and obtained using BepiPred linear epitope prediction method [43].
Antigenicity, Allergenicity and Toxicity Prediction of the Predicted Epitopes
Several bioinformatics studies perform toxicity or allergenicity prediction on peptide candidates to rule out adverse effects in the resulting candidate vaccine [44,45]. In the research of [46] for vaccine development against Chikungunya and Mayaro viruses indicated that the need of the three webservers. The VaxiJen, and ToxinPred webservers were applied consecutively to find out the most appropriate antigenic epitopes, the allergenic and toxic activity of the short-listed B cell and T-cell epitopes respectively [31,47,48]. As the research work of [42] showed, the epitopes that showed high antigenicity, non-allergenicity, non-toxicity, were considered as the best selected epitopes for further analysis and vaccine construction.
Conservancy Analysis of the Selected Epitopes
Conservation analysis is a common method for predicting residues that are functionally important in protein sequences [49]. When designing a vaccine, epitopes of that remain conserved across various strains, are given much priority than genomic regions that are highly variable among the strains since the conserved epitopes of protein(s) provide broader protection across various strains and sometimes even species [50].
The conservancy analysis of the selected epitopes can be performed via the epitope conservancy analysis tool of IEDB server [51]. As the study done by [42] revield, for the conservancy analysis of the DENV-1 epitopes, the envelope protein E from other types of Dengue viruses, DENV- 2, 3 and 4 (UniProt accession numbers: Q8BE39, Q66394, Q7TGD1, Q7TGC7, respectively), were used for comparison that gave 100% conserved sequences which were used for vaccine construction, so the constructed vaccines might also confer immunity towards the DENV serotypes- 2, 3 and 4 along with DENV-1 [52].
Cluster Analysis of the Major Histocompatibility Complex Alleles
Cluster analysis of the Major Histocompatibility Complex (MHC) alleles helps to identify the alleles of the MHC class-I and class-II molecules that have similar binding specificities [53]. In the research work of [42] done on RV approach against DENV-1 and HPV-16 indicated, the cluster analysis of the possible MHC class-I and MHC class-II alleles that may interact with the predicted epitopes were carried out by the online tool MHC cluster 2.0 that can generate the clusters of the alleles in a phylogenetic manner.
Generation of the 3D Structures of the Selected Epitopes
Commonly used software to view 3D structure data are Cn3D available from the national center for biotechnology information (NCBI) website, and Python Language Molecule (PyMOL) and Raster of a Molecule (RasMOL) which are powerful molecular graphics visualization tools with capabilities to highlight segments of user’s interest. Nowadays, numerous tools are available in the new bioinformatics era, and more tools and servers are constantly being added. It is up to the user to make the best use of the available data, information and analytics to derive the end results desired [54,55] used the tools PSI-blast based secondary structure Prediction (PSIPRED) v4.01 and RaptorX-Property [56] to predict the secondary structure of the proposed vaccine and to demonstrate a graphical presentation of it. PSIPRED, a highly accurate predictive secondary structure tool, is a sequence profile-based fold recognition system [55].
As [57] revealed, the 3D structures of the selected best epitopes to identify potential vaccine candidates against SARS-CoV-2 were generated using online 3D generating tool Peptide Fold 3 (PEP-FOLD3) (http://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/) [58].
Molecular Docking of the Selected Epitopes
Molecular docking is an in-silico approach which aims to evaluate the binding affinity between a receptor molecule and a ligand [59]. All together for the appropriate elicitation of immune response, the interaction amongst the antigenic molecule and immune receptor molecule is essential [60]. As the research finding by [27] on RV approach against RSV showed, molecular docking of the epitopes binding to specific human leukocytes antigen (HLA) alleles was performed to evaluate their binding effectiveness [58].
Reverse vaccinology approach to design a novel multi-epitope subunit vaccine against avian influenza A (H7N9) virus by [61] used AutoDOCKVina program to conduct docking based on parameters all the analyses were done at 1.00- A spacing, and the exhaustiveness parameter was kept at 8.00 while the number of outputs was set at 10. All the output Protein Data Bank, Partial Charge (Q), & Atom Type (T) (PDBQT) files were converted in Protein Data Bank (PDB) format using OpenBabel (version 2.3.1). The best output was selected on the basis of lower binding energy. The docking interaction was visualized with the python molecular (PyMOL) graphics system, version 1.5.0.4 (https://www.pymol.org/) [41].
Another research revealed that Toll-like receptor 4 (TLR4) was chosen as the receptor and found from the research collaboratory for structural bioinformatics protein data bank (RCSB PDB) database (https://www.rcsb.org/) (PDB ID: 4G8A) [63] while the vaccine model was used as a ligand. Then, using the ClusPro 2.0 server (https://cluspro.bu.edu/login.php) the binding affinity between the multi-epitope vaccine and the TLR4 receptor was identified via molecular docking [64].
Vaccine Construction
Peptides are the promising candidates as immunotherapeutics, but are less immunogenic when used alone in vaccine design. The peptides need potent immunostimulatory adjuvants to effectively activate the innate and adaptive immunity [66]. As the research work of [67] showed, for vaccine construction, the best selected Cytotoxic T Lymphocyte (CTL); Helper T Lymphocyte (HTL) and B cell Lymphocyte (BCL) epitopes were combined and joined together by suitable linkers [68].
[10] constructed a vaccine against Ebola Virus in sequential manner: first the adjuvant sequence was conjugated with the Pedigree-Aware Distant-Relationship Estimation (PADRE) sequence by EAAAK linker, then the PADRE sequence was added to the CTL epitopes by GGGS linkers, the CTL epitopes were also conjugated with each other by the GGGS linkers. Next, the HTL epitopes were conjugated by GPGPG linkers and the BCL epitopes were linked by KK linkers. The research conducted by [61] on RV approach to design vaccine against Avian Influenza A (H7N9) virus revealed three vaccine sequence were constructed named V1, 236 V2 and V3, each associated with different adjuvants including beta defensin (a 45 mer peptide), 237 L7/L12 ribosomal protein and HABA protein (M. tuberculosis, accession number: AGV15514.1) [68].
Conclusion and Recommendation
Using a reverse vaccinology approach, it is possible to develop effective vaccines against viral diseases in a matter of years and at a low cost without the requirement to culture the organism. Reverse vaccinology against viral diseases employs the entire proteome of the virus by getting it from an online database (e.g., the Uniprot database in the FASTA format). After obtaining the necessary data, epitope selection, prediction, and analysis can be carried out using several bioinformatics tools (which are also publicly available online) in order to produce the right vaccine.
Based on the above conclusion the following recommendations are forwarded:
• Reverse vaccinology approach should be used to design vaccines that are effective against viruses for new, emerging viral diseases that need immediate measurement;
• Due to its rapidity reverse vaccinology approach should be used to overcome challenges of viruses that show genetic diversity; and
Flexible analysis that cannot be performed using conventional methods should be conducted using reverse vaccinology approach.
References
- World Health Organization. Global vaccine action plan 2011–2020. WHO. 2013.
- Robbins JB, Schneerson R, Szu SC, Fattom A, Yang Y, Lagergard T, et al. Prevention of invasive bacterial diseases by immunization with polysaccharide–protein conjugates. Curr Top Microbiol Immunol. 1989; 146: 169–180.
- Hatherill M, White RG, Hawn TR. Clinical development of new TB vaccines: recent advances and next steps. Front. Microbiol. 2019; 10: 3154.
- Sieber CC, Kiesswetter E, Kwetkat A, Heppner HJ, Schoene D, Freiberger E. Prevention: public healthcare, nutrition, physical activity, vaccination. In: Roller-Wirnsberger R., Singler K. and Polidori M.C. editors. Learning geriatric medicine: a study guide for medical students. Cham, Switzerland: Springer International Publishing. 2018; 237–62.
- Wolf MC, Freiberg AN, Zhang T, Akyol-Ataman Z, Grock A, Hong PW, et al. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc Natl Acad Sci USA. 2010; 107: 3157–62.
- Rappuoli R, Aderem A. A 2020 Vision for vaccines against HIV, tuberculosis and malaria. Nature. 2011; 473: 463.
- Sinha S, Kuo CY, Ho JK, White PJ, Jazayeri JA, Pouton CW. A suicidal strain of listeria monocytogenes is effective as a DNA vaccine delivery system for oral administration. Vaccine. 2017; 35: 5115–22.
- Rappuoli R. Reverse vaccinology. Curr Opin Microbiol. 2005; 3: 445-50.
- Borrow R, Balmer P, Miller E. Meningococcal surrogates of protection—serum bactericidal antibody activity. 2005; 23: 2222-2227.
- Ullah A, Sarkar B, Islam SS. Exploiting the Reverse Vaccinology Approach to Design Novel Subunit Vaccine against Ebola Virus. Immunology. 2020; 225: 151949.
- Gupta E, Gupta SR, Niraj RRK. Identification of drug and vaccine target in mycobacterium leprae: a reverse vaccinology approach. International Journal of Peptide Research and Therapeutics. 2019; 1-14.
- Michalik M, Djahanshiri B, Leo JC, Linke D. Reverse Vaccinology: The Pathway from Genomes and Epitope Predictions to Tailored Recombinant Vaccines. Methods in Molecular Biology. 2016; 1403: 87–106.
- Milstien JB, Kaddar M, Kieny MP. The Impact of Globalization on Vaccine Development And Availability. Health Aff (Millwood). 2015; 25: 4.
- Etienne CF. Expanded program on immunization in the Americas: 40 years. Rev. 2017.
- Siegrist CA. Vaccine immunology. In: Plotkin SA, Orenstein WA, Offit PA, editors. Vaccines. 6th ed. Philadelphia, United States: Elsevier/Saunders. 2013; 14–32.
- Delany I, Rappuoli R, De Gregorio E. Vaccines for the 21st century. EMBO Mol Med. 2014; 6: 708–720.
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015; 520: 692–96.
- Alessandro S, Rino R. Review: Reverse Vaccinology: Developing Vaccines in the Era of Genomics. Immunity. 2012; 33: 530-541.
- Bagnoli F, Baudner B, Mishra RP, Bartolini E, Fiaschi L, Mariotti P, et al. Designing the next generation of vaccines for global public health. Omics. 2011; 15: 545–566.
- Monterrubio-López GP, González-Y-Merchand JA, Ribas-Aparicio RM. Identification of novel potential vaccine candidates against tuberculosis Based on reverse vaccinology. BioMed Research International. 2015.
- Usman AA, Saman S, Muhammad SM, Matloob A, Nazia N, Rashid B. Rational design of multi epitope-based subunit vaccine by exploring MERS-COV proteome: Reverse vaccinology and molecular docking approach. PLoS One. 2021.
- Srivastava S, Kamthania M, Singh S, Saxena AK, Sharma N. Structural basis of development of multi-epitope vaccine against Middle East respiratory syndrome using in silico approach. 2018; 11: 2377.
- Yang Y, Zhang L, Geng H, Deng Y, Huang B, Guo Y, et al. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. 2013; 4: 951–961.
- Narula A, Pandey RK, Khatoon N, Mishra A, Prajapati VK. Excavating chikungunya genome to design B and T cell multi-epitope subunit vaccine using comprehensive immunoinformatics approach to control Chikungunya infection. 2018; 61: 4–15.
- Ashfaq UA, Saleem S, Masoud MS, Ahmad M, Nahid N, Bhatti R, et al. Rational design of multi epitope-based subunit vaccine by exploring MERS-COV proteome: Reverse vaccinology and molecular docking approach. Multiepitope based subunit vaccine against MERS-COV. PLoS ONE. 2021; 16: e0245072.
- De Groot AS, Moise L, McMurry JA. Epitope-Based Immunome-Derived Vaccines: A Strategy for Improved Design and Safety. Clinical Applications of Immunomics. 2009; 2: 39–69.
- Qamar MT, Shokat Z, Muneer I, Ashfaq UA, Javed H, Anwar F, et al. Multiepitope-Based Subunit Vaccine Design and Evaluation against Respiratory Syncytial Virus Using Reverse Vaccinology Approach. Vaccines. 2020; 8: 288.
- Hoover DM, Wu Z, Tucker K, Lu W, Lubkowski J. Antimicrobial characterization of human β-defensin 3 derivatives. Antimicrob Agents Chemo ther. 2003; 47: 2804–9.
- Chen W, Anton LC, Bennink JR. 2000 Dissecting the multifactorial causes of immunodominance in class I-restricted T cell responses to viruses. Immunity. 2000; 12: 83–93.
- Kardani K, Bolhassani A, Namvar A. An overview of in silico vaccine design against different pathogens and cancer. Expert Rev. Vaccines. 2020; 19: 699–726.
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinf. 2007; 8: 4.
- Edison O, Mei UW, Anthony H, Yongqun H. COVID-19 coronavirus vaccine design using reverse vaccinology and machine learning. Frontiers in Immunology. 2020; 11: 1581.
- He Y, Xiang Z, Mobley HLT. Vaxign: The first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J. Biomed. Biotechnol. 2010; 2010: 297505.
- Ong E, Wang H, Wong MU, Seetharaman M, Valdez N, He Y. Vaxign-ML: Supervised Machine Learning Reverse Vaccinology Model for Improved Prediction of Bacterial Protective Antigens. Bioinformatics. 2020; 36: 3185-3191.
- Zhao P, Cao J, Zhao LJ, Qin ZL, Ke JS, Pan W, et al. Immune responses against SARS-coronavirus nucleocapsid protein induced by DNA vaccine. Virology. 2005; 5: 331: 128-35.
- Zhou p, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020; 579: 270–273.
- Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020; 395: 565–574.
- Sievers F, Higgins DG. Russell D.J. (ed.) multiple sequence alignment methods. Methods Mol Biol. 2014; 1079: 105-116.
- Krogh A, Larsson B, Von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes 1. J Mol Biol. 2001; 305: 567-580.
- Hajighahramani N, Nezafat N, Eslami M, Negahdaripour M, Rahmatabadi SS, Ghasemi Y. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. 2017; 48: 83–94.
- Kolaskar A, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS letters. 1990; 276: 172-4.
- Sarkar B, Ullah A, Araf Y. A systematic and reverse vaccinology approach to design novel subunit vaccines against Dengue virus type-1 (DENV-1) and human Papillomavirus-16 (HPV-16). Informatics in Medicine Unlocked. 2020.
- Moller S, Croning MD, Apweiler R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 2001; 17: 646–53.
- Alam A, Ali S, Ahamad S, Malik MZ, Ishrat R. From ZikV genome to vaccine: In silico approach for the epitope-based peptide vaccine against Zika virus envelope glycoprotein. Immunology. 2016; 149: 386-399.
- Hajighahramani N, Nezafat N, Eslami M, Negahdaripour M, Rahmatabadi SS, Ghasemi Y. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infection, Genetics and Evolution. 2016; 48: 83-94.
- Hammadul H, Rahatul I, Srijon G, M.d. Mashiur R, Nurnabi AJ, Abunasar M. Implementation of in silico methods to predict common epitopes for vaccine development against Chikungunya and Mayaro viruses. Heliyon. 2021; 7: e06396.
- Gupta S. In silico approach for predicting toxicity of peptides and proteins. PloS One. 2013; 8.
- Dimitrov I. AllergenFP: allergenicity prediction by descriptor fingerprints. 2014; 30: 846–851.
- Capra JA, Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007; 23: 1875-82.
- Bui HH, Sidney J, Li W, Fusseder N, Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinformatics. 2007; 8: 361.
- Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, et al. The immune epitope database (IEDB): 2018 update. Nucleic acids research. 2019; 47: 339-343.
- Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2018; 47: 339–43.
- Thomsen M, Lundegaard C, Buus S, Lund O, Nielsen M. MHC cluster, a method for functional clustering of MHC molecules. Immunogenetics. 2013; 65: 655-65.
- Sarkar T, Das S, De A, Nandy P, Chattopadhyay S, Chawla-Sarkar M, et al. H7N9 influenza outbreak in China 2013: In silico analyses of conserved segments of the hemagglutinin as a basis for the selection of peptide vaccine targets. Computational Biology and Chemistry. 2015; 59: 8–15.
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000; 16: 404-5.
- Wang S, Li W, Liu S, Xu J. RaptorX-Property: a web server for protein structure property prediction. Nucleic Acids Res. 2016; 44: 430-5.
- Gupta E, Mishra RK, Niraj RRK. Identification of potential vaccine candidates against SARS-CoV-2, a step forward to fight novel coronavirus 2019-nCoV: A Reverse Vaccinology Approach. 2020.
- Lamiable A, Thévenet P, Rey J, Vavrusa M, Derreumaux P, Tufféry P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016; 44: 449–454.
- Solanki V, Tiwari V. Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacter baumannii. Scientific reports. 2018; 8: 9044.
- Tovchigrechko A, Vakser IA. GRAMM-X public web server for protein–protein docking. Nucleic Acids Res. 2006; 34: 310–4.
- Hasan M, Ghosh PP, Azim KF, Mukta S, Abir RA, Nahar J, et al. Reverse vaccinology approach to design a novel multi-epitope subunit vaccine against avian influenza A (H7N9) virus. Microb Pathog. 2018.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein Data Bank. Nucleic Acids Res. 2000; 28: 235-42.
- Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, et al. The ClusPro web server for protein-protein docking. Nat Protoc. 2017; 12: 255-278.
- Jones HG, Ritschel T, Pascual G, Brakenho JP, Keogh E, Furmanova-Hollenstein P, et al. Structural basis for recognition of the central conserved region of RSV G by neutralizing human antibodies. PLoS Pathog. 2018; 14: e1006935.
- Nagpal G, Usmani SS, Raghava GP. A web resource for designing subunit vaccine against major pathogenic species of bacteria. Frontiers in Immunology. 2018; 2: 2280.
- Islam R, Parvez A, Anwar S, Hosen MJ. Delineating blueprint of an epitope-based peptide vaccine against the multiple serovars of Dengue virus: A hierarchical reverse vaccinology approach. Informatics in medicine unlocked. 2020.
- Reginald K, Chan Y, Plebanski M, Poh CL. Development of peptide vaccines in Dengue. Curr Pharmaceut Des. 2018; 24: 1157–73.
- Rana A, Akhter Y. A multi-subunit based, thermodynamically stable model vaccine using combined immunoinformatics and protein structure based approach. Immunobiology. 2016; 221: 544–557.