
Review Article
Austin J Vet Sci & Anim Husb. 2024; 11(3): 1146.
Review on Viral Metagenomics as Powerful Tool in Veterinary Medicine
Tewodros Legesse*
Addis Ababa University College of Veterinary Medicine and Agriculture, Ethiopia
*Corresponding author: Tewodros Legesse Addis Ababa University College of Veterinary Medicine and Agriculture, Ethiopia. Email: tewoderos.legesse@aau.edu.et
Received: March 28, 2024 Accepted: May 01, 2024 Published: May 08, 2024
Summary
New diseases continue to emerge in both human and animal populations, and the importance of animals, as reservoirs for viruses that can cause zoonosis are evident. Due to intensive globalization on, climatic changes, and viral evolution, among other factors, the emergence of viruses and new viral diseases has increased in the last decades. Thus, an increased knowledge of the viral flora in animals, both in healthy and diseased individuals, is important both for animal and human health. In this situation, it is crucial to apply powerful methods for the broad-range detection and identification of the emerging viruses. In combination with classical methods, the molecular based techniques provide sensitive and rapid means of virus detection and identification. However, most of the conventional diagnostic tests are designed to be virus specific or aimed at a limited group of infectious agents. In contrast, the novel viral metagenomics approaches allow unbiased detection of a very wide range of infectious agents in a culture independent manner. There are different possible steps of a viral metagenomics study utilizing sequence-independent amplifi cation, high- throughput sequencing, and bioinformatics to identify viruses. The objective of this review is to importance of the applications of viral metagenomics in veterinary science and discusses some of the viruses discovered within this fi eld.
Keywords: Bioinformatics; High-throughput sequencing; Sequence-independent amplification; Viral metagenomics
Abbreviations: cDNA: Complementary DNA; DNA: Deoxyribose Nuclear Acid; DNase: Deoxy-Ribonuclease; dsDNA: Double Stranded DNA; NGS: Next Generation Sequencing; PCR: Polymerase Chain Reaction; RNA: Ribosomal Ribonucleic Acid; RNase: Ribonuclease; rPCR: Random Polymerase Chain Reaction; SISPA: Sequence-Independent Single-Primer Amplification; ssDNA: Single Stranded DNA
Introduction
Throughout recorded history, viruses have caused diseases in animals and humans, with descriptions of possible viral infections appearing long before viruses were fi rst discovered. However, the concept of a virus (meaning poison or toxic) was introduced in 1898 by Beijerinck, who found that the agent causing tobacco mosaic disease in plants could not, as bacteria, be fi ltered out and could not grow by itself but required living and growing cells in which to replicate [7]. They can infect a wide range of hosts including plants, bacteria, fungi, algae, protozoa, vertebrate or non-vertebrate animals. In nature, around 1 × 1031 numbers of different viruses are present. The number it-self suggests the diversity of viruses in nature [35].
The abundance of viruses in nature is around 1000 times more than observed through cell culture dependent technique suggesting that the large pool of viruses is still unknown, only around 219 viral species (belonging to 23 families) that are known to infect humans, among which more than two-thirds are of zoonotic origin [10]. New diseases continue to emerge in both human and animal populations, and the importance of animals, as reservoirs for viruses that can cause zoonosis are evident. Because of this, an increased knowledge of the viral flora in animals, both in healthy and diseased individuals, is important both for animal and human health [15].
Thus, an improved detection of newly emerging and re-emerging viruses and a systematic characterization of the full range of viruses that infect humans are needed [4]. In this situation, it is crucial to apply powerful methods for the broad-range detection and identification of the emerging viruses. In combination with classical methods, the molecular-based techniques provide sensitive and rapid means of virus detection and identification [8]. However, most of the conventional diagnostic tests are designed to be virus-specific or aimed at a limited group of infectious agents. This makes them unsuitable for the detection of unexpected and/or completely new viruses, as well as novel viral variants [8]. In contrast, viral metagenomics approaches allow unbiased detection of a very wide range of infectious agents in a culture-independent manner and novel and highly divergent viruses can be discovered and genetically characterized for the fi rst time [14]. This technology, hold the promise to significantly improve diagnosis and disease control, in line with the ‘‘One World, One Health’’ principles [8].
Therefore, the objective of this seminar is:
• To review metagenomics as a powerful tool for the application of viral metagenomics in veterinary science and discuss some of the viruses discovered within this fi eld.
History of Viral Metagenomics
The genomic age began in 1977 when FX174, a virus that infects Escherichia coli, was sequenced [35]. The metagenomics of viruses began in 2002 with the publication of two uncultured marine viral communities [19]. Mya Breitbart, Forest Rohwer, and colleagues used environmental shotgun sequencing to show that 200 liters of seawater contains over 5000 different viruses [19]. More than 65% of the sequences could not be identifi ed, but a large portion of the identifi ed sequences was from phages, covering most of the major dsDNA phage families [19]. Since then, other studies have been performed, including the viral RNA world [20]. Similar techniques have also been used to study the viral population in various environments, such as feces [18], blood [16] and potential viral reservoirs (e.g., bats) [48]. Because these techniques investigate the complete viral community within a sample, it is possible to study both the natural viral flora and emerging pathogens in disease complexes.
Even though historically it was first associated with the study of uncultured microbial organisms (bacteria and archaea) in environmental samples [37], more recently, it has also been applied to the characterization of viral communities, a task that it is particularly suited because the small size of viral genomes makes their coverage more comprehensive using the same number of metagenomic sequences. Today, viruses are considered the most abundant and diverse living forms on earth [20,72]. Their diversity has been explored by metagenomics in a wide variety of environments: oceans [37], stromatolites [24], acidic hot springs and subterranean and hypersaline environments [24]. The first metagenomic surveys performed on environmental viral communities showed that more than 60% of the sequences had no significant similarity to sequences stored in public databases [16]. This listed benefi t is critical for developing countries to participate to the metagenomics race [15], in particular because of the applications to genomic medicine by implemen ting molecular diagnostics and molecular epidemiology. Science and technology, in particular the life sciences, are increasingly recognized as vital components for national progress in developing countries (Virgin and Todd, 2011).
Workflow of Viral Metagenomics
In the laboratory space, viral metagenomics consists of four major steps: (1) viral enrichment to minimize background of prokaryotic and eukaryotic nucleic acids thus increasing the relative proportion of viral nucleic acids, (2) amplifi cation of viral nucleic acids, (3) sequencing with or without cloning of amplifi ed polymerase chain reaction (PCR) fragments, and (4) bioinformatics analysis of the resulting sequence output [42].
Viral Enrichment
For accurate results, sample collection, proper handling, transportation, stage of the sample is very crucial. There are many standard protocols available for collection of diferent samples to laboratory and its storage techniques. Diferent protocols are used for fluid sample and for tissue samples. The tissue sample is generally homogenized in autoclaved saline and collected supernatant fi ltered through 0.8, 0.45 and 0.2 μm liters, this serial fi ltration procedure is used to separate larger particles and bacteria from viruses. If necessary, the fi rst step of sample procession is homogenization (physical, e.g., homogenizer, mortar and pestle, freeze-thaw cycles; or enzymatic techniques, e.g., salt solution, detergents, alkaline lysis), centrifugation of the sample and fi ltration of the supernatant (through 0.22 and 0.45 μm pore membranes) to remove non-viral nucleic acids (i.e., host cellular debris and bacteria). The fi ltrates can be treated with a mixture of DNases and RNases to further reduce background nucleic acids originating from the host cells and bacteria. The method chosen in this step depends on the physical properties and other characteristics of the sample type [21,54].
Following sample homogenization and reduction of the amount of debris and background nucleic acids, viral particles can be concentrated at various efficacies. Commonly used methods include tangential-flow fi ltration, poly-ethylene glycol precipitation, and ultracentrifugation. Density gradient ultracentrifugation using Cesium Chloride (CsCl) gradient provides highly purifi ed virus particles. Concerning the step of viral nucleic acid extraction, the picture is more complex. Viral particles are disrupted by using “lysis buffer,” which may contain chaotropic acids (e.g., guanidine hydrochloride), detergents (sodium dodecyl sulfate, Triton X-100), and/or proteases (e.g., proteinase K). During the subsequent separation phase, the nucleic acids could be isolated from other components. This can be done by liquid-liquid extraction or liquid-solid extraction.
During the liquid phase extraction different types of alcohol are used (e.g., phenol-chloroform- isoamyl alcohol, isopropanol, etc.). The solid phase extraction may include one of the following procedures: gel fi ltration, where nucleic acid is separated through gel- matrix (e.g., Sephadex), ion exchange chromatography (e.g., anion exchange resin, DEAE-C), and affinity chromatography (silica surface, paramagnetic beads). In general, liquid-solid extraction methods use less hazardous chemicals and provide increased throughput. Various formats have been marketed providing flexible, fast, and scalable viral nucleic acid extraction [21,55,75].
Nucleic Acid Amplification
Amplifi cation of the nucleic acids is performed in a sequence-independent manner to show the true genetic composition of the sample. This step is able to simultaneously multiply several viral genomes including highly divergent and completely novel viruses and thus enable their discovery and genetic characterization [3,9,23]. Some of the more commonly used sequence-independent strategies are briefly described in the following sections as well as a number of different sequencing technologies.
Sequence-independent single-primer amplification: The ligation of adaptors to cDNA and DNA, either directly or after restriction digestion, enables sequence-independent amplifi cation of all nucleic acids in a sample. Two decades ago, a version of this strategy, called sequence-independent single-primer amplifi cation (SISPA) [64], was shown in combination with immunoscreening to retrieve and aid in the genetic characterization of a Norwalk virus in fecal samples [51]. This strategy and modifi cations to it, such as DNase-SISPA [2] and VIDISCA [79], have successfully been used to identify a number of viruses, such as new Anello- and Parvoviruses in humans [41] and Bungowannah virus in pigs with Porcine Myocarditis (PMC) syndrome [44].
Random PCR: An alternative amplifi cation technology is random PCR (rPCR), which neither requires the digestion of the DNA/cDNA nor the ligation of adaptors. The rPCR utilizes a primer that consists of a known adaptor sequence at the 5-prime end followed by a degenerate hexa- or heptamer at the 3-prime end. Using this primer in the cDNA synthesis step, the cDNA is labeled with the adaptor sequence at both ends (a similar step can be performed on the DNA), which enables the creation of primer target sites for the subsequent PCR reaction. This strategy has been widely used for the investigation of viral metagenomes, and a number of novel viruses have been discovered using this strategy in combination with sequencing [30].
Displacement amplification: Another efficient amplifi cation strategy is the use of random primers in combination with a displacement polymerase [40]. Phi29 is a high-fi delity displacement polymerase with high processivity that can incorporate over 70,000 bases prior to detaching from its template [12,27], and only a few nanograms of starting material can produce several micrograms of DNA in one reaction. Using random primers and phi29, circular targets are efficiently amplifi ed through a reaction termed ‘‘rolling-circle amplifi cation,’’ which creates long concatemers of multiple copies of the target [22].
Circular targets are ideal for this amplifi cation strategy. However, linear targets can also be amplifi ed in a similar manner [22]. For short linear DNA viruses and cDNA, amplifi cation is less efficient, and additional steps, such as ligation, may have to be included for amplifi cation (Berthet et al., 2008). This strategy has also been successful in studying different viral metagenomes, and several novel viruses have been discovered by this method, such as a fi bropapilloma virus in sea turtles [55], Anellovirus in harbor seals [56], Bocavirus in pigs [15], and Papillomaviruses in humans [63].
Sequencing Technologies
Owing to efforts in research and innovation (Figure 3), sequencing capacity and speed have dramatically increased over the past decade, while the cost is continuously decreasing. These achievements together led to the capacity to produce billions of nucleotide bases in a single sequencing run, which was unconceivable some time ago [26].
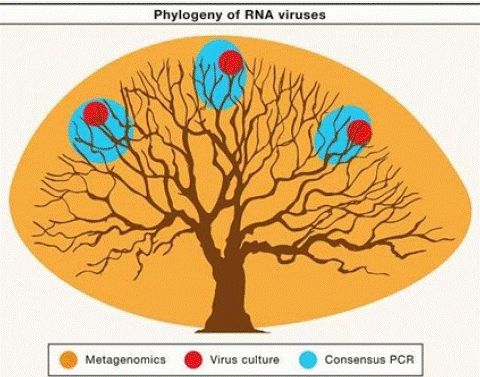
Figure 1: Different Ways to Explore the Virosphere: A tree is used to represent the total phylogenic diversity of RNA viruses. The colored circles illustrate the extent of this diversity that can be discovered using three common approaches for virus discovery: cell culture, consensus PCR, and metagenomics.
Source: (Zhang et al., [84]).
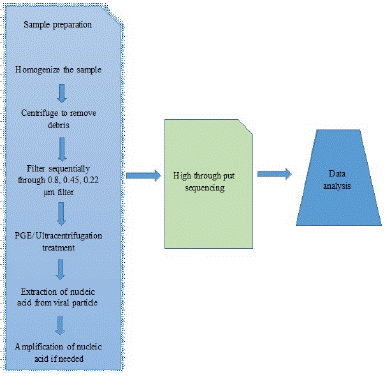
Figure 2: Overview of general procedures of metagenomics.
Source: (Bhukya and Nawadkar, [10]).
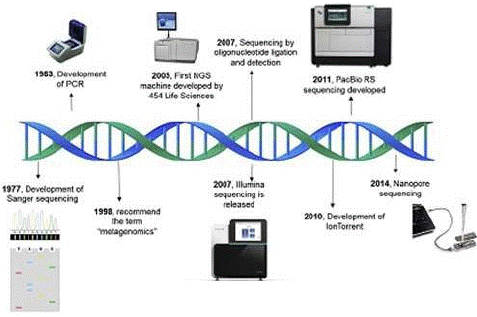
Figure 3: Metagenomics timeline and milestones: Timeline showing advances in sequencing technologies from Sanger sequencing to Nanopore sequencing.
Source: (Kaszab et al., [42]).
![]()
Animal species
Type studies
Animal viruses
(Viral reads taxonomic assignation)Zoonosis
Pigs
DNA/RNA virome (serum)
Asfariviridae, Anelloviridae, Retroviridae
Togaviridae (Alphavirus)
DNA/RNA virome (stool)
Picornaviridae, stroviridae, Caliciviridae
Not documented
Bush Pigs
DNA/RNA virome (serum)
Parvoviridae, Circoviridae, Retroviridae
Not documented
Source: (Temmam et al., [74]).
Table 1: Examples of virus detected in Suid species by metagenomic diversity studies.
To identify the viral nucleic acid in a sample, sequencing is often utilized. One approach is to construct viral shotgun libraries and sequence these [16] by a standard sequencing technology such as for example Sanger sequencing (Sanger et al., 1977). This approach creates high quality sequence data and can nowadays produce sequence reads of up to almost 1000 nucleotide (nt). But as this approach is highly laborious in comparison to its yield, the use of new high-throughput sequencing technologies is often replacing Sanger sequencing for metagenomic studies. High throughput sequencing omits the need for bacterial cloning and produces greater amounts of data compared to Sanger sequencing [43,52].
For viral metagenomics, this enables the detection of viruses with low copy numbers. And even though a variety of methods are used to reduce the host and other contaminating nucleic acids, a vast amount still remains and has the potential of masking the viral nucleic acids. Therefore, high- throughput sequencing is often required in the viral metagenomics screening although Sanger sequencing is still often used in the follow-up studies due to its capacity to produce longer sequencing reads [14].
High-throughput sequencing: In 2005, the 454 platform by Roche was the fi rst commercial high-throughput sequencing technology to be released [49]. In the following years, two other platforms (Solexa/Illumina and the SOLiD system) also became commercially available. Both the clonal amplifi cation and the sequencing strategy differ between these three technologies, as do the amount of data produced and the read length. Both 454 and SOLiD utilize emulsion PCR for the clonal amplifi cation, while the Solexa/Illumina system uses bridge PCR. As mentioned before, the actual sequencing chemistry also differs between these platforms [43,52]. The 454 platform is based on pyrosequencing and while it produces the longest reads (400 nt) compared to the others, its throughput is less (0.4–0.6 Gb/run). Solexa/Illumina uses a system with reversible terminators and has a higher throughput (3–6 Gb/run) with a read length of 100 nt. The highest throughput has SOLiD (10–20 Gb/run), which is based on ligation and cleavable probes, but the reading length is only 50 nt [60].
There are benefi ts and drawbacks to these sequencing technologies that should be considered when choosing between the different platforms. For viral metagenomics studies, the 454 platform is widely used because of the longer read lengths, which facilitate de novo assembly. However, Solexa/Illumina has also been used for this purpose. For example, by analyzing a Solexa/Illumina dataset originating from small RNAs isolated from mosquitoes, several viruses were found [83]. New platforms are also being introduced, one being Ion Torrent (Life Technologies), which is based on the release of hydrogen ions as nucleotides are being incorporated by the polymerase [60].
Although the above-mentioned sequencing technologies do not require bacterial cloning, they all still require each product to be clonally amplifi ed prior to sequencing. However, there are new single-molecule sequencing technologies in development that do not require this step [60] and in 2007, Helicos BioSciences released the fi rst single-molecule sequencing platform [34]. Single-molecule sequencing can be an advantage as it avoids errors that are introduced by the pre-sequencing amplifi cation and it will also give a truer picture regarding the frequencies of different DNA fragments. In addition to the above-mentioned advantages, some of the technologies being developed also show promise in obtaining substantial longer read lengths. They are based on alternative sequencing strategies such as: (1) nanopore sequencing in which DNA is threaded through a nanopore and each base recorded as it moves through the pore or (2) sequencing by synthesis in which polymerases are immobilized and the incorporation of nucleotides into a DNA molecule is recorded [34].
Bioinformatics of Viral Metagenomics
One of the challenges in viral metagenomics can be found in the analysis of the vast amount of sequencing data produced. Unlike re-sequencing of viral genomes with high-throughput sequencing by which it is possible to map the reads into an existing genome, the datasets from metagenomic studies are complicated by the fact that they contain a mixture of different species. Bioinformatic analyses of viral metagenomes attempts to answer three questions: how many viruses are there (diversity), ‘what are they (taxonomy), and what are they doing (function)? [80].
Also, the genomes in the datasets are usually incomplete with some cases wherein there are only a few numbers of short fragments belonging to each genome. Furthermore, some reads display a high divergence compared to sequences that are deposited in databases [82]. For these reasons, de novo assembly can be a difficult task, and many of the existing de novo assemblers have been developed for the longer reads produced by Sanger sequencing. However, a number of de novo assembly algorithms are being evaluated for their suitability to this specifi c task, and more are sure to be developed [5,53,62]. After assembly, many researchers rely on the use of programs, such as Blastn® and Blastx®, for database searches to fi nd homologies to known viruses. Although these methods have been used to detect known viruses as well as highly divergent viruses, they often fail to discover completely novel viruses because they do not show similarity to anything deposited in the databases.
Most metagenomics datasets include a number of sequences that cannot be annotated through homology searches in databases. To fi nd novel viruses, other tools have to be developed. Toward this end, many researchers are investigating if it is possible to use motifs, such as di and tri- nucleotide frequencies, codon usage, and secondary structures, to distinguish viral sequences [23,78]. Several programs and platforms have been developed such as, MEGAN® [38,39] PathSeq [45] and CAMERA® [5,69] that can help with both the analysis of the data as well as with the visualization of the results.
Follow-up of Findings and Causations
Depending on the purpose of the study and on the viral discovery, the results of a viral metagenomic study can be followed up in a variety of different ways. Generally, the complete genomes of the discovered viruses are not obtained through current viral metagenomic procedures, and further genetic characterization for complete genome characterization is required. Virus isolation, molecular and genetic characterization, design of diagnostic tools, prevalence studies, experimental infections, etc., can be used for a better understanding of the role of the discovered virus(es) [47]. If the study aims to determine the etiology behind a disease, it is important to remember that the detection of nucleic acid using these techniques is not sufficient to prove causality [29]. To prove causality, more comprehensive studies have to be performed. However, viral metagenomics can help to guide the direction of the investigation [14].
Applicat?i on of Viral Metagenomics
Application of viral metagenomics is being continuously adopted and used in veterinary medicine. In clinical diagnostics, the recognition and the treatment of novel and rare pathogens is a real challenge. With the new technical inventions, this problem could be solved. Another application is the real-time investigation of outbreaks caused by viral pathogens and the prevention of potential epidemics. The environmental monitoring of pathogens by viral metagenomics may also have an important role in efforts of infectious disease control. In this case, the quick reaction, the regulation, and risk assessment based on laboratory confi rmed evidence is very important. Moreover, broad- range detection and identifi cation of novel viruses or novel types of known viruses will be able to help us to broaden our range of vision, facilitate protection against viral agents, and understand viral diversity [42].
Viral Discovery
In former times, the discovery of new viruses or novel virus variants happened mainly as the consequence of isolation on cell culture, embryonated chicken eggs, or in animal models. Virus growth was detected due to disease development, cytopathic effects, and the use of broad diagnostics like electron microscopy or antisera for neutralization or staining. Detection of completely new viruses was often by accident only, depending, e.g., on the possibility to grow the virus.
Novel DNA sequencing techniques, known as Next-Generation Sequencing? (NGS) techniques, are new tools providing high-throughput sequence data with many possible applications in research and diagnostic settings. With the development of different NGS platforms, it is now possible to sequence all viral genomes in a given sample without previous knowledge about their nature with the use of sequence-independent amplifi cation followed by high-throughput sequencing. This combination of techniques, known as viral metagenomics, allows the discovery of completely new viral species within a complex sample and, due to decreasing costs, is nowadays exponentially increasing [6]. A growing number of examples exists now where viruses were detected in samples from diseased animals or in animal reservoirs [36].
Diagnostics
Clinical diagnostics is perhaps the most desirable developmental goal of viral metagenomic pathogen detection. In many cases, clinical diagnoses of viral infections require tedious culturing and diagnostic tests; however, for rare or novel pathogens these tests may be insufficient or in conclusive, and even identification of common pathogens remains challenging [73]. Additionally, the difficulty associated with identifying viral pathogens may lead to improper clinical treatment, such as the administration of antibiotics [61]. The unambiguous and target-independent identification of all viral pathogens in a relevant clinical sample would help clinicians to direct treatment properly and avoid misdiagnoses. When applied in clinical samples, this approach also has the ability to identify coinfections, some of which may previously have been overlooked after the diagnosis of an initial infection. Although technical developments are necessary to deploy viral metagenomics as a stand-alone diagnostic approach, the disruptive potential of viral metagenomics is immense and has great potential to shift the way we conduct clinical diagnoses of viral infections [11].
Outbreak Response
Another application for which metagenomics is well suited is the detection and response to viral pathogen outbreaks [65]. Wildlife often act as a reservoir for viruses in nature, and as the interface between wildlife, humans and domestic animals increases due to encroachment, these viruses have the potential to spill-over and cause diseases and/or death in the new hosts (Daszak et al., 2000). Viral metagenomics studies have demonstrated the abundance and divergence of viruses present in bats, and these studies have identifi ed bats as important viral carriers of known and currently unknown viruses. Approximately, 72% of all emerging zoonotic diseases are estimated to have a wildlife reservoir, including SARS and Hantavirus pulmonary disease (Feldmann et al., 2002; Jones et al., 2008).
Another sub-application is for detection of pathogens deployed in bioterrorism attacks. Pathogenic viral strains may be either naturally or engineered to be divergent from known relatives, hindering traditional diagnostics. Synthetic genes inserted into a viral host result in both a more infectious agent and an agent that can escape detection by traditional methods. For example, in the case of a novel wild type Ebola virus outbreak, not all traditional tests identified Ebola virus; however, the metagenomic approach identified the causative agent in all cases. Through a monitoring network enabled by viral metagenomics, previously termed ‘Public Health Metagenome Surveillance’, we will be better suited to defend against and respond to all disease outbreaks, both natural and human- made [54].
Environmental Monitoring
In complex environmental samples such as sewage-polluted water, the potential for pathogen diversity is high. Typically, areas such as recreational beaches are monitored for concentrations of fecal coliform bacteria; however, it has been long recognized that these bacteria are not accurate indicators of viral pathogens [31]. Pathogen diversity is likely high in environments affected by pollution. Target-independent metagenomic viral pathogen detection has the potential to direct research efforts, risk assessment, and regulation more effectively at pathogens that are highly enriched in the environment [81]. Initial surveys have indeed shown high and unexpected pathogen diversity, with identification of emerging viruses being far more prevalent than for viruses that are typically monitored. Identifying actual pathogen presence and diversity through metagenomics, rather than solely the presence of indicator organisms, represents a significant improvement over current environmental monitoring practices and regulation [66].
Food Safety
The incidence and impact of foodborne illness constitutes a signifi cant global issue to public health. It is another fi eld for which diagnostic metagenomics can improve pathogen detection. Foodborne pathogenic bacteria like Salmonella, Listeria, or toxin-producing Escherichia coli strains, but also norovirus (65%) and Hepatitis A virus and parasites (Trichinella, Giardia) can cause disease out- breaks accompanied by vomiting and more or less severe diarrhea [76]. Metagenomics for example offers the advantage of a less biased pathogen detection methodology through direct sequencing of the specimen’s extracted DNA. This approach has the potential to capture a thorough representation of the microbial community. A number of clinically relevant applications stand to benefi t from such data, rapid identifi cation of the etiological agent (known or novel) and gene content including virulence and AMR, or inferring functional pathways to elucidate multifaceted illnesses. Thus, routinely applied for foodborne pathogens, resulting metagenomics data can be helpful for surveillance as well as foodborne outbreak investigation and will improve the hazard identifi cation by increased specifi city and potentially by a fundamental change in the defi nition of the hazard being rather a specifi c virulent strain, subtype, or gene instead of a not well-specifi ed species [70].
Basic Research in (veterinary) Virology
While the aforementioned examples illustrate the impact of high-throughput sequencing on veterinary diagnostics and the closely connected fi eld of food safety, there are of course diverse applications beyond. Researchers only recently started using shotgun high-throughput sequencing to systematically assess the pathogen content of historic samples available in museums or other collections and ancient DNA from archaeologic specimens. Whereas anthropologists already successfully analyzed ancient samples immediately after the early second-generation sequencers became available [32,50,58].
Systematic investigation of historic samples will not only help discover the diversity of the virome but will essentially contribute to the elucidation of viral evolution and the potential impact of vaccination on the evolving virome. At the moment, however, no reports on the analysis of historic animal samples have been published, but the studies of historic DNA samples are focused to human samples. Duggan and colleague sequenced a complete variola virus genome from a 17th century child mummy from Lithuania [25]. The authors were able to reconstruct the complete viral genome, which was found to be basal to all strains from the 20th century. The authors concluded that much of variola virus evolution and diversifi cation occurred recently driven by the impact of vaccination [25]. Another study published shed further light on the long-term evolution of variola virus. In their study, they report on the sequencing and analysis of two complete variola virus genomes from historic human specimens from a museum in Prague. Together these two studies using shotgun high-throughput sequencing provide a new level of insight into long-term virus evolution [59].
Challenges and Future Aspects of Viral Metagenomics
Challenges of Viral Metagenomics
Metagenomics has quickly become a major tool for exploring viral diversity, yet several challenges need to be addressed in order to fully leverage the potential of these methods. First, metagenomes built from limited input material are still difficult to reliably obtain and interpret, and do not yet provide a comprehensive and quantitative view of the viral community present in the sample [67].
The second major challenge lies in the absence of direct host information for genomes assembled from metagenomes. In a clinical context, this means that one of Koch’s postulates, which requires that the candidate etiological agent be isolated from a diseased organism and grown in pure culture, cannot be fulfilled. A modified Koch’s postulate for the metagenomics era has been proposed in which potential new pathogens first must be present and more abundant in the diseased subject compared to matched control. Then, experiments using either a sample from a disease subject or an artificial virus obtained through DNA synthesis and expression in cell cultures must be performed to demonstrate that this agent induces disease in another healthy subject [67]. Still, we lack clear knowledge about the link between the diversity of virus in the environment and during outbreaks, our surveillance is merely based on a biased collection of only clinical samples and their study. This limits our knowledge about disease spread.
Prediction of future outbreaks and limiting the spread of disease needs proper study, development of strong tool [57].
Future Aspects
The amazing diversity and novelty of viral metagenomes mean that large-scale sequencing efforts like the acid mine drainage and Sargasso Sea projects need to be carried out on the viral component. These surveys will provide the raw data necessary for understanding the size of the viral metagenome and community structure. Methods to clone and sequence ssDNA and RNA viruses also need to be developed and incorporated into these surveys to include all viruses in these analyses. At the bioinformatics level, tools need to be automated and made freely available so individual labs can carry out viral metagenomics analyses on communities of interest [16].
Conclusion and Recommendation
In summary, viral metagenomics and the tools for the analysis of the data produced will continue to be developed and refi ned with rapid improvement in the genomic sequencing techniques. The overall metagenomics approach is very valuable for discovery of new viruses, novel genes, surveillance of pathogens, discover new pathway, host virus interaction, functional studies. These listed benefi ts are critical for developing countries to participate to the metagenomics race. The leads obtained through this exercise may have great impact on early diagnosis and treatment. While metagenomics studies also experience limitations and challenges, which need to overcome in near future to obtain a precise result. Unifi ed genomic extraction techniques and development of improved analysis modules may suffice the needs of metagenomics in future.
• Collaborative research must be promoted to advance developing countries in the early detection of the causative agent for preliminary preparation using metagenomics so that emerging and re-emerging diseases won’t pose threats.
• Viral metagenomics can solve problems associated with clinical interventions, like treatment of animal diseases which fail due to a limited knowledge about the viral agents, so supporting and funding research involving viral metagenomics can enhance animal’s health which indirectly enhancing public health.
References
- Alavandi SV, Poornima M. Viral metagenomics: a tool for virus discovery and diversity in aquaculture. Indian J Virol. 2012; 23: 88-98.
- Allander T, Emerson SU, Engle RE, Purcell RH, Bukh J. A virus discovery method incorporating DNase treatment and its application to the identifi cation of two bovine parvovirus species. In: Proceedings of the National Academy of Sciences. 2001; 98: 11609–11614.
- Ambrose HE, Clewley JP. Virus discovery by sequence-independent genome amplifi cation. Rev Med Virol Virol. 2006; 16: 365–83.
- Anderson NG, Gerin JL, Anderson NL. Global screening for human viral pathogens. Emerging Infect Dis. 2003; 9: 768–774.
- Bao S, Jiang R, Kwan W, Wang B, Ma X, Song YQ. Evaluation of next-generation sequencing software in mapping and assembly. J Hum Genet. 2011; 56: 406–414.
- Barzon L, Lavezzo E, Militello V, Toppo S, Palù G. Applications of next-generation sequencing technologies to diagnostic virology. Int J Mol Sci. 2011; 12: 7861–7884.
- Beijerinck MW. Concerning a contagium viwm fluidum as cause of the spot disease of tobacco leaves. Phytopathology Classics. 1898; 7: 33-52.
- Belak S, Karlsson OE, Blomstrom AL, Berg M, Granberg F. New viruses in veterinary medicine, detected by metagenomic approaches. Vet Microbiol. 2013; 165: 95-101.
- Bexfield N, Kellam P. Metagenomics and the molecular identifi cation of novel viruses. Vet J. 2011; 190: 191-198.
- Bhukya PL, Nawadkar R. Potential applications and challenges of metagenomics in human viral infections. Metagenomics for Gut Microbes. 2018; 9: 19.
- Bibby K. Metagenomic identifi cation of viral pathogens. Trends Biotechnol. 2013; 31: 275-279.
- Blanco L, Bernad A, Lazaro JM, Martin G, Garmendia C, Salas M. Highly efficient DNA synthesis by the phage phi 29 DNA polymerase. Symmetrical mode of DNAreplication. J Biol Chem. 1989; 264: 8935–8940.
- Blomstrom AL, Widen F, Hammer AS, Belak S, Berg M. Detection of a novel astrovirus in brain tissue of mink suffering from shaking mink syndrome by use of viral metagenomics. J Clin Microbiol. 2010; 48: 4392–6.
- Blomstrom AL. Viral metagenomics as an emerging and powerful tool in veterinary medicine. Vet Q. 2011; 31: 107-114.
- Blomström AL, Belák S, Fossum C, McKillen J, Allan G, Wallgren P, et al. Detection of a novel porcine boca-like virus in the background of porcine circovirus type 2 induced postweaning multisystemic wasting syndrome. Virus Res. 2009; 146: 125-129.
- Breitbart M, Rohwer F. Method for discovering novel DNA viruses in blood using viral particle selection and shotgun sequencing. Biotechniques. 2005; 39: 729-36.
- Breitbart M, Felts B, Kelley S, Mahaffy JM, Nulton J, Salamon P, et al. Diversity and population structure of a near-shore marine-sediment viral community. Proc Biol Sci. 2004; 271: 565–574.
- Breitbart M, Hewson I, Felts B, Mahaffy JM, Nulton J, Salamon P, et al. Metagenomic analyses of an uncultured viral community from human feces. J Bacteriol. 2003; 185: 6220-6623.
- Breitbart M, Salamon P, Andresen B, Mahaffy JM, Segall AM, Mead D, et al. Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci. 2002; 99: 14250–14255.
- Culley AI, Lang AS, Suttle CA. Metagenomic analysis of coastal RNA virus communities. Science. 2006; 312: 1795–1798.
- Datta S, Budhauliya R, Das B, Chatterjee S, Hmuaka V, Veer V. Next-generation sequencing in clinical virology: discovery of new viruses. World J Virol. 2015; 4: 265-276.
- Dean FB, Nelson JR, Giesler TL, Lasken RS. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primedmultiply primed rolling circle amplification. Genome Res. 2001; 1: 1095–9.
- Delwart EL. Viral metagenomics. Rev Med Virol. 2007; 17: 115–31.
- Desnues C, Rodriguez-Brito B, Rayhawk S, Kellev S, Trati T, Havnes M, et al. Biodiversity and biogeography of phages in modern stromatolites and thromatolites. Nature. 2008; 452: 340–3.
- Duggan AT, Perdomo MF, Piombino-Mascali D, Marciniak S, Poinar D, Emery MV. 17th century variola virus reveals the recent history of small pox. Curr Biol. 2016; 26: 3407–3412.
- Escobar-Zepeda A, Vera-Ponce De Leon A, Sanchez-Flores A. The road to metaMeta- genomics: from microbiology to DNA sequencing technologies and bioinformatics. Front Genet. 2015; 6: 348.
- Esteban JA, Salas M, Blanco L. Fidelity of phi 29 DNA polymerase. Comparison between protein-primed initiation and DNA polymerization. J Biol Chem. 1993; 268: 2719–2726.
- Feldman M, Harbeck M, Keller M, Spyrou MA, Rott A, Trautmann B, et al. A high -coverage Yersinia pestis genome from a sixth -century justinianic plague victim. Mol Biol Evol. 2016; 33: 2911–2923.
- Fredericks DN, Relman DA. Sequence-based identifi cation of microbial pathogens: a reconsideration of Koch’s postulates. Clin Microbiol Rev. 1996; 9: 18–33.
- Froussard P. A random-PCR method (rPCR) to construct whole cDNA library from low amounts of RNA. Nucleic Acids Res. 1992; 20: 2900.
- Gerba CP, Goyal SM, LaBelle RL, Cech I, Bodgan GF. Failure of indicator bacteria to reflect the occurrence of enteroviruses in marine waters. Am J Public Health. 1979; 69: 1116–1119.
- Green RE, Krause J, Ptak SE, Briggs AW, Ronan MT, Simons JF. Analysis of one million base pairs of Neanderthal DNA. Nature. 2006; 444: 330–336.
- Hanke D, Pohlmann A, Sauter-Louis C, Hoper D, Stadler J, Ritzmann M. Porcine epidemic diarrhea in Europe: In -detail analyses of disease dynamics and molecular epidemiology. Viruses. 2017; 9: 177.
- Harris TD, Buzby PR, Babcock H, Beer E, Bowers J, Braslavsky I, et al. Single-molecule DNA sequencing of a viral genome. Science. 2008; 320: 106–109.
- Hobbie JE, Daley RJ, Jasper S. Use of nuclepore ilters for counting bacteria by fluorescence microscopy. Appl Env Microbio. 1977; 33: 1225-1228.
- Höper D, Wylezich C, Beer M. Loeffler 4.0: Diagnostic Metagenomics. In Advances in virus research. Academic Press. 2017; 99: 17-37.
- Hugenholtz P, Tyson GW. Microbiology: metagenomics. Nature. 2008; 455: 481–483.
- Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomics data. Genome Res. 2007; 17: 377–386.
- Huson DH, Richter DC, Mitra S, Auch AF, Schuster SC. Methods for comparative metagenomics. BMC Bioinformatics. 2009; 10: S12.
- Johne R, Muller H, Rector A, van Ranst M, Stevens H. Rolling-circle amplification of viral DNA genomes using phi29 polymerase. Trends Microbiol. 2009; 17: 205–11.
- Jones MS, Kapoor A, Lukashov VV, Simmonds P, Hecht F, Delwart E. New DNA viruses identified in patients with acute viral infection syndrome. J Virol. 2005; 79: 8230–8236.
- Kaszab E, Doszpoly A, Lanave G, Verma A, Bányai K, Malik YS, et al. Metagenomics revealing new virus species in farm and pet animals and aquaculture. In Genomics and Biotechnological Advances in Veterinary, Poultry, and Fisheries. 2020; 29-73.
- Kircher M, Kelso J. High-throughput DNA sequencing–concepts and limitations. Bioessays. 2010; 32: 524-536.
- Kirkland PD, Frost MJ, Finlaison DS, King KR, Ridpath JF, Gu X. Identification of a novel virus in pigs-Bungowannah virus: a possible new species of pestivirus. Virus Res. 2007; 129: 26–34.
- Kostic AD, Ojesina AI, Pedamallu CS, Jung J, Verhaak RG, Getz G, et al. PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat Biotechnol. 2011; 29: 393–396.
- Krause L, Diaz NN, Goesmann A, Kelley S, Nattkemper TW, Rohwer F, et al. Phylogenetic classification of short environmental DNA fragments. Nucleic Acids Res. 2008; 36: 2230–2239.
- Leland DS, Ginocchio CC. Role of cell culture for virus detection in the age of technology. Clin Microbiol. Rev. 2007; 20: 49–78.
- Li L, Victoria JG, Wang C, Jones M, Fellers GM, Kunz TH, et al. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J Virol. 2010; 84: 6955-6965.
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in micro-fabricated high-density picolitre reactors. Nature. 2005; 437: 376–380.
- Maricic T, Paabo S. Optimization of 454 sequencing library preparation from small amounts of DNA permits sequence determination of both DNA strands. Biotechniques. 2009; 46: 51–57.
- Matsui SM, Kim JP, Greenberg HB, Su W, Sun Q, Johnson PC, et al. The isolation and characterization of a Norwalk virus - specifi c cDNA. J Clin Invest. 1991; 87: 1456–1461.
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010; 11: 31– 46.
- Miller JR, Koren S, Sutton G. Assembly algorithms for next-generation sequencing data. Genomics. 2010; 95: 315–327.
- Mokili JL, Rohwer F, Dutilh BE. Metagenomics and future perspectives in virus discovery. Curr Opin Virol. 2012; 2: 63-77.
- Ng TF, Manire C, Borrowman K, Langer T, Ehrhart L, Breitbart M. Discovery of a novel single-stranded DNA virus from a sea turtle fi bropapillomafi bro-papilloma by using viral metagenomics. J Virol. 2009; 83: 2500–2509.
- Ng TF, Wheeler E, Greig D, Waltzek TB, Gulland F, Breitbart M. Metagenomic identifi cation of a novel anellovirus in Pacifi c harbor seal (Phoca vitulina richard- sii) lung samples and its detection in samples from multiple years. J Gen Virol. 2011; 92: 1318–1323.
- Nieuwenhuijse DF, Koopmans MP. Metagenomic sequencing for surveillance of food-and waterborne viral diseases. Front Microbiol. 2017; 8: 230.
- Noonan JP, Coop G, Kudaravalli S, Smith D, Krause J, Alessi J. Sequencing and analysis of Neanderthal genomic DNA. Science. 2006; 314: 1113–1118.
- Pajer P, Dresler J, Kabickova H, Pisa L, Aganov P, Fucik K. Characterization of two historic smallpox specimens from a Czech museum. Viruses. 2017; 9: 200.
- Pareek CS, Smoczynski R, Tretyn A. Sequencing technologies and genome sequencing. J Appl Gen. 2011; 52: 413-435.
- Pavia AT. Viral infections of the lower respiratory tract: old viruses, new viruses, and the role of diagnosis. Clin Infect Dis. 2011; 52: 284–289.
- Pop M. Genome assembly reborn: recent computational challenges. Brief Bioinform. 2009; 10: 354–366.
- Rector A, Bossart GD, Ghim SJ, Sundberg JP, Jenson AB, Van Ranst M. Characterization of a novel close- to-root papillomavirus from a Florida manatee by using multiply primed rolling-circle amplifi cation: trichechus manatus latirostris papillomavirus type 1. J Virol. 2004; 78: 12698–12702.
- Reyes GR, Kim JP. Sequence-independent, single-primer amplifi cation (SISPA) of complex DNA populations. Mol Cell Probes. 1991; 5: 473-481.
- Rosario K, Breitbart M. Exploring the viral world through metagenomics. Curr Opin Virol. 2011; 1: 289–297.
- Rosario K, Nilsson C, Lim YW, Ruan Y, Breitbart M. Metagenomic analysis of viruses in reclaimed water. Environ Microbio. 2009; 11: 2806-2820.
- Roux S, Matthijnssens J, Dutilh BE. Metagenomics in Virology. Reference Module in Life Sciences. 2019; 1–8.
- Salim AF, Phillips AD, Farthing MJ. Pathogenesis of gut virus infection. Baillière’s Clinical Gastroenterology. 1990; 4: 593-607.
- Seshadri R, Kravitz SA, Smarr L, Gilna P, Frazier M. CAMERA: a community resource for metagenomics. PLoS Biol. 2007; 5: e75.
- Stasiewicz MJ, den Bakker HC, Wiedmann M. Genomics tools in microbial food safety. Curr Opin Food Sci. 2015; 4: 105–110.
- Sun S, Chen J, Li W, Altintas I, Lin A, Peltier S, et al. Community cyberinfrastructure for Advanced Microbial Ecology Research and Analysis: the CAMERA resource. Nucleic Acids Res. 2011; 39: 546–551.
- Suttle CA. Viruses in the sea. Nature. 2005; 437: 356–361.
- Talbot HK, Falsey AR. The diagnosis of viral respiratory disease in older adults. Clin Infect Dis. 2010; 50: 747–751.
- Temmam S, Davoust B, Berenger JM, Raoult D, Desnues C. Viral metagenomics on animals as a tool for the detection of zoonoses prior to human infection?. Int J Mol Sci. 2014; 15: 10377–10397.
- Thatcher SA. DNA/RNA preparation for molecular detection. Clin Chem. 2015; 61: 89-99.
- Thomas MK, Murray R, Flockhart L, Pintar K, Fazil A, Nesbitt A, et al. Estimates of foodborne illness–related hospitalizations and deaths in Canada for 30 specifi ed pathogens and unspecifi ed agents. Foodborne Pathog Dis. 2015; 12: 820-827.
- Toppinen M, Perdomo MF, Palo JU, Simmonds P, Lycett SJ, Soderlund-Venermo M. Bones hold the key to DNA virus history and epidemiology. Sci Rep. 2015; 5: 17226.
- Trifonov V, Rabadan R. Frequency analysis techniques for identifi cation of viral genetic data. MBio. 2010; 1: e00156-10.
- Van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJ, Wolthers KC, et al. Identifi cation of a new human coronavirus. Nat Med. 2004; 10: 368–373.
- Willner DL. Viral metagenomics in host-associated systems (Doctoral dissertation, UC San Diego). 2010.
- Wong K, Fong TT, Bibby K, Molina M. Application of enteric viruses for fecal pollution source tracking in environmental waters. Environ Int. 2012; 45: 151-164.
- Wooley JC, Godzik A, Friedberg I. A primer on metagenomics. PLoS Comput Biol. 2010; 6: e1000667.
- Wu Q, Luo Y, Lu R, Lau N, Lai EC, Li WX, et al. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. In Proceedings of the National Academy of Sciences. USA. 2010; 107: 1606–1611.
- Zhang YZ, Shi M, Holmes EC. Using metagenomics to characterize an expanding virosphere. Cell. 2018; 172: 1168-1172.