
Review Article
Austin Virol and Retrovirology. 2014;1(2): 6.
TIM-3, a Possible Target for Immunotherapy in Cancer and Chronic Viral Infections
Roger Tieu1, Praveen Kumar Amancha1, François Villinger1,2 and Siddappa N. Byrareddy2*
1Division of Microbiology and Immunology, Yerkes National Primate Research Center, USA
2Department of Pathology & Laboratory Medicine, Emory University School of Medicine, USA
*Corresponding author: Siddappa N. Byrareddy, Department of Pathology & Laboratory Medicine, Emory University School of Medicine, 101 Woodruff Circle, WMRB, Room 2337A, Atlanta, GA 30322
Received: November 06, 2014; Accepted: December 03, 2014; Published: December 05, 2014
Abstract
Effector T-cell responses are controlled by complex mechanisms involving various soluble factors and co-stimulatory and co-inhibitory molecules. These inhibitory receptors act as immune checkpoints and are extensively investigated as possible therapeutic targets, such as PD-1 and CTLA-4. Recently TIM-3 is also gaining prominence in tumor and chronic viral infection models as a candidate for immunotherapy in conjunction with other inhibitory receptors. This review discusses the recent findings on the expression of TIM-3 and its ligand in tumor and chronic viral infection.
Keywords: TIM-3; CTLA-4; Immunotherapy; Galectin-9
Introduction
Upon exposure to non-self entities such as pathogens, a number of receptors including Pattern Recognition Receptors (PRRs) are triggered leading to a rapid response of the innate immune system [1]. This initial response paves the way for the elaboration of pathogen specific adaptive humoral and cell mediated responses via a complex network of co-stimulatory and inhibitory signals. The initial efferent response by and large favors co-stimulatory signaling for the activation and proliferation of effector cells with the intent of eliminating the invading pathogen. After controlling or eliminating the threat, the absence of continuing antigen stimulation leads to elimination of most effector cells via apoptosis while a fraction of these cells become long term memory cells and homeostasis is reestablished. However, during the course of cancer and chronic persisting viral infections, this return to normalcy is perturbed leading to immune dysfunction and dysregulation resulting in the continued presence of effector cells with poor function. The mechanisms leading to such muted phenotype are increasingly being characterized and involve the expression of receptors, which are thought to play a central role in this inhibition of function.
These inhibitory mechanisms are an integral and critical contributor to immune homeostasis, in minimizing collateral damage during extended inflammatory response as well as preventing autoimmunity by maintaining peripheral self-tolerance. Such pathways serve to restrain effector cell activity, mainly of T cells. Inappropriately activated lymphocytes recognizing self-antigen subsequently lose effector function and undergo apoptosis or energy. Such responses require constricted regulation and transient expression of inhibitory signaling. Prolonged and/or over expression of multiple inhibitory receptors have been associated with impaired immune function such as T cell exhaustion, a state characterized by poor effector function, a mechanism that is exploited by many persistent pathogens.
During chronic active infections, a progressive loss of function in pathogen-specific CD8+ T cells was documented following a hierarchy with an initial impaired production of IL-2 and cytotoxicity, followed by loss of TNF-α, and eventually IFN-γ at late stages [2], resulting in a condition known as exhaustion [2,3] (Figure 1). T cell exhaustion has been found to be associated with the up-regulation of inhibitory receptors in concert with their ligands also up-regulated on Antigen Presenting Cells (APCs) [3]. The initial marker associated with inhibition of function has been Cytotoxic T Lymphocyte Associated Antigen-4 (CTLA-4), a marker upregulated on the surface of activated T cells, with markedly higher affinity for CD80/CD86, the co-stimulatory ligands to CD28 during the elaboration of an antigen specific response [4]. More recently, PD-1 (programmed death 1) was found to be selectively up-regulated by exhausted T cells during murine chronic LCMV (Lymphocytic Choriomeningitis Virus) infection, and blocking of this receptor with antibodies enhanced virus-specific CD8+ T cell response and decreased viral load [5]. This finding defined a novel role for inhibitory pathway involvement in controlling T cell response in chronic viral infection as well as the potential for targeting these same pathways. However, blockade of the PD-1 pathway only partially restored T cell functions, suggesting the involvement of other inhibitory pathways. Additional inhibitory receptors have been since identified which include T cell immunoglobulin and mucin domain 3 (TIM-3), CD244 (2B4), killer Cell Lectin like Receptor G1 (KLRG1), and Lymphocyte Activation Gene 3 (LAG3) [6-12].
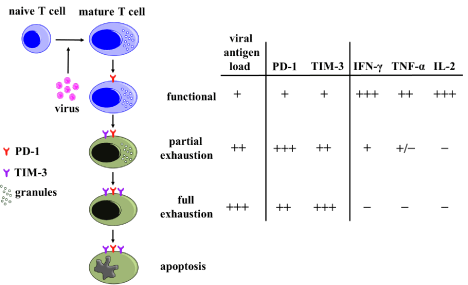
Figure 1: A proposed model of T cell exhaustion in chronic viral infection. As duration of infection and antigen presence increase, T cells progressively lose the ability to produce cytokines, proliferate, and survive. PD-1 acts early in T cell exhaustion upon T cell activation, whereas TIM-3 acts later. IFN-γ, TNF-α, and IL-2 represent markers for immune activation. The concentration of each entity is presented on a scale from high (+++) to low (–) [3].
TIM-3, a receptor involved in the regulation of immune response
TIM-3 is a type I transmembrane protein consisting of an N-terminal Immunoglobulin Variable (IgV)-like domain, a mucin domain with potential sites of O- and N-linked glycosylation, followed by a transmembrane domain, and a cytoplasmic tail with tyrosine phosphorylation motifs. The murine TIM-family genes were cloned from a segment of chromosome 11 homologous to human 5q23-35 associated with asthma susceptibility [13]. Polymorphisms in TIM-3 have been associated with development of airway hyper-reactivity in murine models and rheumatoid arthritis in humans [13,14] while defects in TIM-3 expression contribute to multiple sclerosis pathology [15].
TIM-3has been reported to be expressed by virus-specific T cells during HIV and HCV infections, and its expression levels were correlated with the degree of T cell exhaustion in a manner akin to PD-1 [16,17] (Figure 1). TIM-3 was initially identified as a marker expressed on T helper 1 (Th1) but not on Th2 cells [18], to help maintain peripheral tolerance through interaction with TIM-3 ligand expressed by regulatory T cells (Tregs) [19,20]. Interaction between TIM-3 and its ligand, galectin-9, negatively regulates Th1 cells through selective deletion of TIM-3+ Th1 cells to control population and activity of Th1 cells [21]. Blockade of TIM-3 in vitro was shown to restore antigen specific proliferation and cytokine production by “exhausted” T cells. The addition of TIM-3 as an inhibitory pathway involved in T cell exhaustion introduced the idea of apoptosis as the final stage for exhausted T cells.
While these studies provided for an interesting and relatively simple paradigm, subsequent studies have reported the expression of TIM-3 on Th17 cells [22,23] Natural Killer (NK) and Natural Killer T (NKT) cells [24-27], dendritic cells [28-30], and macrophages/ monocytes [18,29-33], suggesting a far more complex role for this molecule, beyond T cell exhaustion. The complex nature of TIM- 3 and its broad expression across the various innate and adaptive effector cells illustrate the fascinating interplay with other regulatory molecules and warrants further investigation.
Galectin-9, a member of endogenous lectin acts as a ligand for TIM-3
Galectin-9 was identified as a ligand binding to carbohydrate motifs on IgV domain of murine TIM-3 [21]. Galectin-9 is an S-type lectin with two distinct carbohydrate recognition domains joined by a linker peptide [34], and can bind to glycans featuring galactose and other sugars. Target carbohydrates recognized by galectin-9 remain to be elucidated but structural analysis has revealed that galectin-9 can complex with poly-N-acetyllactosamine-containing structures [35]. Various tissues including the thymus express galectin-9 [34]. Given the nature of TIM-3/galectin-9 interactions, much effort has focused on identifying a specific galectin-9 expressing population. Initial reports have found Tregs and naïve CD4+ T cells to express galectin-9 though additional studies will be needed to elucidate in what context galectin-9 assumes an active role [36,37]. Interestingly, one study showed a lack of interaction between galectin-9 and TIM- 3 from both murine and human origin [38]. However, it must be noted that this study utilized membrane-bound galectin-9 rather than the soluble form of galectin-9 used in the other studies, and such a difference in methodology may impact signaling and partly explain the discrepancies. Furthermore, structural analysis of TIM-3 revealed galectin-9 independent binding sites for the IgV region [39]. Tetramers of bacterially expressed TIM-3, which lack any glycosylation, bound to unknown ligand(s) on T cells, B cells, DCs, and macrophages. These studies warrant the investigation of additional ligands that may interact with the full-length TIM-3 protein, or its IgVor mucin domains, which have yet to be explored.
Stimulation of TIM-3 expressing T cells with galectin-9 resulted in decreased susceptibility to HIV infection, via downregulation of HIV-1 co-receptors, CCR5, CXCR4 and α4β7 on the surface of activated CD4+ T cells [40]. In HIV-infected T cells, the presence of galectin-9 limits viral replication. Conversely, resting T cells lacking TIM-3 expression exhibited increased HIV infection in the presence of galectin-9 in a PD1-dependent manner. A different study demonstrated the expression of TIM family proteins, including TIM- 3, enhances HIV infection of CD4+ T cells but limits viral replication and release of virus from infected cells in an apoptotic-independent manner [41]. However, another study showed galectin-9 not as a pro-apoptotic agent, but rather as an inducer of pro-inflammatory cytokines from T helper cells in a TIM-3 independent manner [42]. In a controversial study, TIM-3 was shown not to interact with galectin-9 and in fact, blockade of TIM-3 signaling in human T cells did not restore effector function [38]. These findings suggest that the interaction of TIM-3 and galectin-9 is likely far more complex than a simple interaction and may heavily depend upon the context of the interaction.
Role of TIM-3 in cancer/tumor models and in chronic viral infections
Attempts to identify new Th1-specific cell surface proteins resulted in the identification of TIM-3 [18]. Treatment with antibody specific to TIM-3 exacerbated Experimental Autoimmune Encephalomyelitis (EAE) in mice [18], and increased the proliferation and activation of macrophages. Although the underlying mechanism was not elucidated, these studies suggested two possible mechanisms; 1) cross-linking of TIM-3 receptors on Th1 cells promotes an increased pro-inflammatory state, which may activate macrophages directly or indirectly; 2) Disrupting interactions between TIM-3 and its inhibitory ligand on macrophages allows for their activation in the presence of (or independent of) Th1 cells.TIM-3 expressing DCs can be either stimulatory or inhibitory depending on the microenvironment. A recent study illustrated that the expression of TIM-3 was markedly upregulated on tumor infiltrating DCs and that such expression correlated with inhibition of PRR mediated innate responses, independent from the effect of galectin-9. Instead, TIM- 3 was shown to bind HMGB1 (high mobility group box protein 1) inhibiting the recruitment of nucleic acids from dying tumor cells as HMGB1/nucleic acid complexes to the endo some and thereby blocking the activation of the DCs [43]. Without proper stimulation of DCs, TIM-3 expressing tumor-specific CD8+ T cells will fail to escape their exhausted state [44]. In mice bearing solid tumors, a large fraction of tumor infiltrating lymphocytes were shown to be co-expressing PD-1 and TIM-3 [45]. This population exhibited a more severe exhausted state and a combined targeting of PD-1 and TIM-3 showed better control of the tumor [45]. Similar findings were noted for the Acute Myelogenous Leukemia (AML) model in mice [46].
Increased expression of TIM-3 has been observed on exhausted CD8+ T cells isolated from individuals infected with Human Immunodeficiency Virus (HIV) [16,47,48], Hepatitis B Virus (HBV) [49,50] and Hepatitis C Virus (HCV) [8,17,47] and this level of TIM- 3 expression correlated with ineffective antiviral responses. Murine models of chronic viral infections have been used to observe altered patterns of effector function of TIM-3 expressing T cells in HBV [51], Herpes Simplex Virus (HSV) [52] and LCMV infection. Of note, one study demonstrated that HIV infection of CD4+ T cells does not induce expression of TIM-3 on T cells [53]. Instead, TIM-3 expression may be the result of immune activation through TCR/CD28 co-simulation and the presence of common γ-chain (γc) cytokines IL-2, IL-7, IL-15, and IL-2, but not IL-4 [53].
Recently, it has been suggested that TIM-3 and PD-1 expressing cells marked distinct populations, with a minority expressing both in HIV-infected individuals [16]. TIM-3 expression levels directly correlated with clinical prognosis and inversely with CD4+ T cell counts. Exhausted CD8+ T cells were characterized by increased expression of TIM-3, but blockade of the TIM-3 pathway restored partial function with increased production of IFN-γ in vitro. TIM- 3 presents itself as a viable therapeutic target as demonstrated by enhanced HIV-specific T cell response. However, it remains unclear whether prolonged blockade of TIM-3 regulation of T cell functions may result in increased inflammation and potentially breaking of tolerance to self.
Resting T cells show minimal expression of TIM-3, which is upregulated only after prolonged stimulation. Unlike resting T cells, resting NK cells constitutively expressed TIM-3 [25,26]. TIM-3 upregulation appeared to mark the transition from CD56brightCD16– NK cells to mature CD56dimCD16+ NK cells [25]. Stimulation of TIM-3 expressing NK cells resulted in cytokine production and cytotoxicity. In a different study, the presence of galectin-9 significantly enhanced the ability of TIM-3 expressing NK cells to produce IFN-γ [26]. However, recent publications have reported conflicting findings regarding the role of TIM-3 on NK cells [27,54]. NK cells isolated from HBV patients had high expression levels of TIM-3, and blockade of TIM-3 signaling with TIM-3 blocking antibodies or soluble TIM-3 (sTIM-3) protein led to increased cytotoxicity and IFN-γ production in vitro, suggesting TIM-3 signaling plays a suppressive role for NK cell effector functions [54]. NK cells from advanced melanoma patients demonstrated an increased upregulation of TIM- 3 and displayed an exhausted phenotype characterized by impaired cytotoxicity, IFN-γ production and cell proliferation [27]. Blockade of TIM-3 with various antibodies promotes internalization of the TIM- 3 receptor and upregulation of the IL-2 Receptor (IL-2R). Increased density of IL-2R on the cell-surface renders NK cells more responsive to IL-2 stimulation, thus improving NK cell-mediated cytotoxicity, suggesting a potential mechanism linked to alternate signaling via the internalized TIM-3. However, similarities to prior studies are also apparent, with decreased NK cell cytotoxicity upon cross linking of TIM-3 by antibody or binding to soluble or membrane-bound galectin-9. Interestingly, the addition of galectin-9 blocking antibody did not affect cytotoxicity, suggesting TIM-3 negatively regulates NK cell function in a galectin-9-independent fashion, thus implicating additionalTIM-3 ligands. It is important to note that the studies portraying TIM-3 as an NK cell activation marker only evaluated healthy donors, whereas the studies implicatingTIM-3 as an NK cell suppression marker evaluated patients with chronic diseases. Such a distinction may play an important role in understanding the modular response of TIM-3 depending on the disease context.
TIM-3: target for immunotherapy
During chronic viral infections with sustained antigen stimulation, exhausted CD8+and CD4+T cells can co-express multiple inhibitory receptors, which can work synergistically to modulate the functional quality of these virus-specific T cells. The severity of the dysfunction has a direct correlation with the combined number of expressed and putatively triggered inhibitory receptors. Co-expression of TIM-3 and PD-1 on CD8+ T cells in a murine model of LCMV was associated with severe CD8+ T cell exhaustion characterized by markedly diminished proliferation and cytokine production upon antigen stimulation [55]. TIM-3 and PD-1 function cooperatively to limit T cell function, since blockade of both pathways was markedly more effective thanTIM-3 blockade or PD-1 blockade alone, which only partially restore T cell effector function. Upon dual blockade, T cell function restoration was significantly greater than blockade either single pathway suggesting additive or even synergy.
However, as alluded to above, TIM-3 blockade like any blockade of natural immune inhibitory mechanism will have to be carefully evaluated in vivo for potential exacerbation of inflammatory responses and the potential for breaking tolerance to self, if used for prolonged periods of time. In that regard, testing in murine models may or may not provide relevant findings for a translation to the clinic.
Future Directions
Many questions pertaining to TIM-3 remain unanswered. At the molecular level, the glycosylation pattern of the TIM-3 receptor remains to be elucidated. Carbohydrate moieties on TIM-3 play an important role in ligand binding as evident by galectin-9 binding the TIM-3 IgV domain in a carbohydrate-dependent manner [21]. Given that the TIM-3 mucin domain also carries carbohydrate moieties for ligand interactions, it would be of considerable interest to determine the role of the mucin domain as well as its potential ligands. The combined finding of a galectin-9 independent binding site on TIM-3 and that non-glycosylated soluble TIM-3 binds to numerous primary immune cells suggests that there are additional ligands involved in the immune regulation of various cell lineages [39]. The identification of an endogenous soluble TIM-3 is another intriguing finding that begs the question of whether this isoform may have a different glycosylation pattern [19].
Future investigations must keep in mind the context in which TIM-3 and soluble TIM-3 is expressed since their respective function appears to markedly vary depending on the disease, environment, and temporal context. Currently, it remains an open question as to whether the TIM-3 expressing T cells from cancer patients represent an equivalent functional population to TIM-3 expressing T cell from HIV infected people. In the light of unexpected and often times conflicting findings, it is important to keep in mind that species specific differences may exist and murine, rhesus macaque, and human biological pathways may not match up [56-58] as described previously (70-72).
Also the longevity of exhausted TIM-3+CD8+T cells needs to be addressed: Thus, are these the same cells that are indeed being rescued or conversely, does blockade of TIM-3 (and other inhibitory markers) prevent newly activated cell from becoming exhausted? These are questions that need to be addressed in the context of immunotherapy aiming to reinvigorate of these populations in vivo. It may be possible to stimulate an effective response from T cells by inducing severe variations in levels of antigen followed by antigen specific stimulation, which is currently being explored with discontinuation of ART and administration of Fc-fusion proteins during SIV-infection [59,60] (Figure 2). Given that cases of spontaneous control of HCV have been reported, it is possible that exhausted T cells may be activated in response to an environmental cue [61]. Additional studies are needed to evaluate these concepts. Furthermore, it is unclear whether the dysfunctional states of CD4+ and CD8+ T cells are comparable. And other primary immune cell lineages including B cells [62], NK cells [27,54], and DCs [63] have been reported to exhibit exhaustion as well, the role of TIM-3 in these conditions remain to be elucidated.

Figure 2: Overview of the immune checkpoint molecules PD-1 and TIM-3. (Left) PD-1 binding to its ligand PD-L1 (or PD-L2) and TIM-3 binding to its ligand galectin-9 (or other) mediate an exhausted state in T cells resulting in decreased proliferative capacity and cytokine production. (Right) Blockade of these interactions usingPD-1-Fc [60], soluble TIM-3 (sTIM-3), and TIM-3-Fc represent a means to rescue T cells, thus restoring proliferative capacity and cytokine production.
Conclusion
Finally, it has become clear that TIM-3 plays an intricate role along with other receptors in balancing immune activation and suppression. Given the great complexity of the crosstalk between the various signaling pathways, computational analysis and systems biology will take upon more of a role in understanding immunological mechanisms and disease pathogenesis [64-67], especially those involving TIM-3. The integration of knowledge from all the “omics” fields, including genomic, transcriptomic, proteomic, metabolomic, and lipidomic, will be essential in dissecting the role of TIM-3.
Acknowledgement
This work was supported in part by grants from the NIH (R01 AI113883) to SN Byrareddy, (R01 AI078775 and 8R24 OD010947) to FVillinger.
References
- Medzhitov R, Janeway C Jr. Innate immunity. N Engl J Med. 2000; 343: 338-344.
- Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. Journal of Virology. 2003; 77: 4911-4927.
- Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009; 138: 30-50.
- Coyle AJ, Gutierrez-Ramos JC. The expanding B7 superfamily: increasing complexity in costimulatory signals regulating T cell function. Nature Immunology. 2001; 2: 203-209.
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006; 439: 682-687.
- Bengsch B, Seigel B, Ruhl M, Timm J, Kuntz M, Blum HE, et al. Coexpression of PD-, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. 2010; 6: e1000947.
- Raziorrouh B, Schraut W, Gerlach T, Nowack D, Grüner NH, Ulsenheimer A, et al. The immunoregulatory role of CD244 in chronic hepatitis B infection and its inhibitory potential on virus-specific CD8+ T-cell function. Hepatology. 2010; 52: 1934-1947.
- McMahan RH, Golden-Mason L, Nishimura MI, McMahon BJ, Kemper M, Allen TM, et al. Tim-3 expression on PD-1+ HCV-specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte-directed in vitro cytotoxicity. J Clin Invest. 2010; 120: 4546-4557.
- Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nature Immunology. 2009; 10: 29-37.
- Nakamoto N, Kaplan DE, Coleclough J, Li Y, Valiga ME, Kaminski M, et al. Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology. 2008; 134: 1927-1937.
- Kaufmann DE, Kavanagh DG, Pereyra F, Zaunders JJ, Mackey EW, Miura T, et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nature Immunology. 2007; 8: 1246-1254.
- Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012; 72: 917-927.
- McIntire JJ, Umetsu SE, Akbari O, Potter M, Kuchroo VK, Barsh GS, et al. Identification of Tapr (an airway hyper reactivity regulatory locus) and the linked Tim gene family. Nature Immunology. 2001; 2: 1109-1116.
- Chae SC, Park YR, Shim SC, Yoon KS, Chung HT. The polymorphisms of Th1 cell surface gene Tim-3 are associated in a Korean population with rheumatoid arthritis. Immunol Lett. 2004; 95: 91-95.
- Koguchi K, Anderson DE, Yang L, O'Connor KC, Kuchroo VK, Hafler DA. Dysregulated T cell expression of TIM3 in multiple sclerosis. J Exp Med. 2006; 203: 1413-1418.
- Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. 2008; 205: 2763-2779.
- Golden-Mason L, Palmer BE, Kassam N, Townshend-Bulson L, Livingston S, McMahon BJ, et al. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol. 2009; 83: 9122-9130.
- Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002; 415: 536-541.
- Sabatos CA, Chakravarti S, Cha E, Schubart A, Sánchez-Fueyo A, Zheng XX, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nature Immunology. 2003; 4: 1102-1110.
- Sánchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nature Immunology. 2003; 4: 1093-1101.
- Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng XX. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005; 6: 1245-1252.
- Seki M, Oomizu S, Sakata KM, Sakata A, Arikawa T, Watanabe K, et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clinical Immunology. 2008; 127: 78-88.
- Hastings WD, Anderson DE, Kassam N, Koguchi K, Greenfield EA, Kent SC, et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. 2009; 39: 2492-2501.
- Khademi M, Illés Z, Gielen AW, Marta M, Takazawa N, Baecher-Allan C, et al. T Cell Ig- and mucin-domain-containing molecule-3 (TIM-3) and TIM-1 molecules are differentially expressed on human Th1 and Th2 cells and in cerebrospinal fluid-derived mononuclear cells in multiple sclerosis. Journal of Immunology. 2004; 172: 7169-7176.
- Ndhlovu LC, Lopez-Vergès S, Barbour JD, Jones RB, Jha AR, Long BR, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. 2012; 119: 3734-3743.
- Gleason MK, Lenvik TR, McCullar V, Felices M, O'Brien MS, Cooley SA, et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood. 2012; 119: 3064-3072.
- da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res. 2014; 2: 410-422.
- Nagahara K, Arikawa T, Oomizu S, Kontani K, Nobumoto A, Tateno H, et al. Galectin-9 increases Tim-3+ dendritic cells and CD8+ T cells and enhances antitumor immunity via galectin-9-Tim-3 interactions. J Immunol. 2008; 181: 7660-7669.
- Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science. 2007; 318: 1141-1143.
- Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009; 113: 3821-3830.
- Zhang Y, Ma CJ, Wang JM, Ji XJ, Wu XY, Jia ZS, et al. Tim-3 negatively regulates IL-12 expression by monocytes in HCV infection. PLoS One. 2011; 6: e19664.
- Zhang Y, Ma CJ, Wang JM, Ji XJ, Wu XY, Moorman JP, et al. Tim-3 regulates pro- and anti-inflammatory cytokine expression in human CD14+ monocytes. J Leukoc Biol. 2012; 91: 189-196.
- Jayaraman P, Sada-Ovalle I, Beladi S, Anderson AC, Dardalhon V, Hotta C, et al. Tim3 binding to galectin-9 stimulates antimicrobial immunity. J Exp Med. 2010; 207: 2343-2354.
- Wada J, Kanwar YS. Identification and characterization of galectin-9, a novel beta-galactoside-binding mammalian lectin. J Biol Chem. 1997; 272: 6078-6086.
- Nagae M, Nishi N, Murata T, Usui T, Nakamura T, Wakatsuki S, et al. Structural analysis of the recognition mechanism of poly-N-acetyllactosamine by the human galectin-9 N-terminal carbohydrate recognition domain. Glycobiology. 2009; 19: 112-117.
- Wang F, Wan L, Zhang C, Zheng X, Li J, Chen ZK. Tim-3-Galectin-9 pathway involves the suppression induced by CD4+CD25+ regulatory T cells. Immunobiology. 2009; 214: 342-349.
- Oomizu S, Arikawa T, Niki T, Kadowaki T, Ueno M, Nishi N, et al. Cell surface galectin-9 expressing Th cells regulate Th17 and Foxp3+ Treg development by galectin-9 secretion. PLoS One. 2012; 7: e48574.
- Leitner J, Rieger A, Pickl WF, Zlabinger G, Grabmeier-Pfistershammer K, Steinberger P. TIM-3 does not act as a receptor for galectin-9. PLoS Pathog. 2013; 9: e1003253.
- Cao E, Zang X, Ramagopal UA, Mukhopadhaya A, Fedorov A, Fedorov E, et al. T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent ligand-binding surface. Immunity. 2007; 26: 311-321.
- Elahi S, Niki T, Hirashima M, Horton H. Galectin-9 binding to Tim-3 renders activated human CD4+ T cells less susceptible to HIV-1 infection. Blood. 2012; 119: 4192-4204.
- Li M, Ablan SD, Miao C, Zheng YM, Fuller MS, Rennert PD, et al. TIM-family proteins inhibit HIV-1 release. Proc Natl Acad Sci U S A. 2014; 111: E3699-3707.
- Su EW, Bi S, Kane LP. Galectin-9 regulates T helper cell function independently of Tim-3. Glycobiology. 2011; 21: 1258-1265.
- Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nature Immunology. 2012; 13: 832-842.
- Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006; 6: 715-727.
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Anderson. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010; 207: 2187-2194.
- Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011; 117: 4501-4510.
- Vali B, Jones RB, Sakhdari A, Sheth PM, Clayton K, Yue FY, et al. HCV-specific T cells in HCV/HIV co-infection show elevated frequencies of dual Tim-3/PD-1 expression that correlate with liver disease progression. Eur J Immunol. 2010; 40: 2493-2505.
- Sakhdari A, Mujib S, Vali B, Yue FY, MacParland S, Clayton K, et al. Tim-3 negatively regulates cytotoxicity in exhausted CD8+ T cells in HIV infection. PLoS One. 2012; 7: e40146.
- Nebbia G, Peppa D, Schurich A, Khanna P, Singh HD, Cheng Y, et al. Upregulation of the Tim-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS One. 2012; 7: e47648.
- Wu W, Shi Y, Li S, Zhang Y, Liu Y, Wu Y, et al. Blockade of Tim-3 signaling restores the virus-specific CD8? T-cell response in patients with chronic hepatitis B. Eur J Immunol. 2012; 42: 1180-1191.
- Ju Y, Hou N, Zhang XN, Zhao D, Liu Y, Wang JJ, et al. Blockade of Tim-3 pathway ameliorates interferon-gamma production from hepatic CD8+ T cells in a mouse model of hepatitis B virus infection. Cell Mol Immunol. 2009; 6: 35-43.
- Sehrawat S, Reddy PB, Rajasagi N, Suryawanshi A, Hirashima M, Rouse BT. Galectin-9/TIM-3 interaction regulates virus-specific primary and memory CD8 T cell response. PLoS Pathog. 2010; 6: e1000882.
- Mujib S, Jones RB, Lo C, Aidarus N, Clayton K, Sakhdari A, et al. Antigen-Independent Induction of Tim-3 Expression on Human T Cells by the Common ?-Chain Cytokines IL-2, IL-7, IL-15, and IL-21 Is Associated with Proliferation and Is Dependent on the Phosphoinositide 3-Kinase Pathway. J Immunol. 2012; 188: 3745-3756.
- Ju Y, Hou N, Meng J, Wang X, Zhang X, Zhao D, et al. T cell immunoglobulin- and mucin-domain-containing molecule-3 (Tim-3) mediates natural killer cell suppression in chronic hepatitis B. J Hepatol. 2010; 52: 322-329.
- Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2010; 107: 14733-14738.
- Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004; 172: 2731-2738.
- Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013; 110: 3507-3512.
- Shay T, Jojic V, Zuk O, Rothamel K, Puyraimond-Zemmour D, Feng T, et al. Conservation and divergence in the transcriptional programs of the human and mouse immune systems. Proc Natl Acad Sci U S A. 2013; 110: 2946-2951.
- Amancha PK, Hong JJ, Rogers K, Ansari AA, Villinger F. In vivo blockade of the programmed cell death-1 pathway using soluble recombinant PD-1-Fc enhances CD4+ and CD8+ T cell responses but has limited clinical benefit. J Immunol. 2013; 191: 6060-6070.
- Finnefrock AC, Tang A, Li F, Freed DC, Feng M, Cox KS, et al. PD-1 blockade in rhesus macaques: impact on chronic infection and prophylactic vaccination. J Immunol. 2009; 182: 980-987.
- Lauer GM, Kim AY. Spontaneous Resolution of Chronic Hepatitis C Virus Infection: Are We Missing Something? Clin Infect Dis. 2006; 42: 953-954.
- Moir S, Ho J, Malaspina A, Wang W, DiPoto AC, O'Shea MA, et al. Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J Exp Med. 2008; 205: 1797-1805.
- Langenkamp A, Messi M, Lanzavecchia A, Sallusto F. Kinetics of dendritic cell activation: impact on priming of TH, TH2 and nonpolarized T cells. Nat Immunol. 2000; 1: 311-316.
- Park KY, Li WA, Platt MO. Patient specific proteolytic activity of monocyte-derived macrophages and osteoclasts predicted with temporal kinase activation states during differentiation. Integr Biol (Camb). 2012; 4: 1459-1469.
- Pulendran B. Systems vaccinology: probing humanity's diverse immune systems with vaccines. Proc Natl Acad Sci U S A. 2014; 111: 12300-12306.
- Rappuoli R, Aderem A. A 2020 vision for vaccines against HIV, tuberculosis and malaria. Nature. 2011; 473: 463-469.
- Burton BR, Britton GJ, Fang H, Verhagen J, Smithers B, Sabatos-Peyton CA, et al. Sequential transcriptional changes dictate safe and effective antigen-specific immunotherapy. Nature Communications 5. 2014.