
Short Communication
Austin J Clin Cardiolog. 2014;1(2): 1011.
Genetics and Mechanisms of Catecholaminergic Polymorphic Ventricular Tachycardia
Hiroshi Watanabe* and Tohru Minamino
Division of Cardiology, Niigata University Graduate School of Medical and Dental Sciences, Japan
*Corresponding author: Hiroshi Watanabe, Division of Cardiology, Niigata University Graduate School of Medical and Dental Sciences, 1-754 Asahimachidori, Niigata, Japan
Received: January 27, 2014; Accepted: February 20, 2014; Published: February 24, 2014
Abstract
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited arrhythmia syndrome characterized by ventricular tachyarrhythmia induced by adrenergic stress in the absence of structural heart disease and high incidence of sudden cardiac death. Five causative genes of CPVT have been identified. There is a common mechanism by which mutations in these genes cause CPVT: Ca2+ leakage through the destabilized ryanodine channel complex in sarcoplasmic reticulum. Spontaneous Ca2+ release through ryanodine channel leads to delayed after depolarization, triggered activity, and bidirectional⁄polymorphic VT. Implantable cardioverter defibrillators (ICDs) are used for prevention of sudden death in patients with CPVT. However, because painful shocks can trigger further adrenergic stress and tachyarrhythmias, ICD socks delivered to initiating triggered arrhythmias have nearly always failed and deaths have occurred despite appropriate ICD shocks. Treatment with β–adrenergic blockers reduces arrhythmia burden and mortality, but is not completely effective. The beneficial effects of Ca2+ channel blocker verapamil in combination with β–blocker have been reported, but the role of verapamil has not been well assessed. Flecainide directly inhibits ryanodine channels and prevent CPVT. Left cardiac sympathetic denervation may be an effective alternative treatment in combination with ICD, especially for patients whose arrhythmias are not controlled by drug therapies. Catheter ablation for premature ventricular beats triggering CPVT has recently been effective.
Keywords: Arrhythmias; Genetics; Mechanism; Ryanodine channel; Therapy
Introduction
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited arrhythmia syndrome characterized by ventricular tachyarrhythmia induced by adrenergic stress in the absence of structural heart disease and high incidence of sudden cardiac death [1–4]. The age of CPVT onset is usually before 10 years, although the much later onset has been reported [4]. The diagnosis is made based on reproducible ventricular tachyarrhythmias including bidirectional VT, which is characterized by a beat–to–beat 180° rotation of the QRS axis and polymorphic VT during exercise testings and⁄or epinephrine administration. Five causative genes of CPVT have been identified (Table 1). There is a common mechanisms by which mutations in these genes cause CPVT: Ca2+ leakage through the destabilized ryanodine channel complex in sarcoplasmic reticulum (Figure 1) [5]. Spontaneous Ca2+ release through ryanodine channel leads to delayed after depolarization, triggered activity, and bidirectional⁄polymorphic VT. Therefore, ryanodine channel block can be therapeutic for CPVT, and it has recently discovered that flecainide directly inhibits ryanodine channels and prevent CPVT [6–8].
![]()
Gene
Protein
Transmission mode
Frequency
RYR2
Ryanodine receptor
Autosomal dominant
60%
CASQ2
Calsequestrin
Autosomal recessive
1-2%
TRDN
Triadin
Autosomal recessive
Rare
KCNJ2
Inward rectifier K+ channel, Kir 2.1
Autosomal dominant
Rare
CALM1
Calmodulin
Autosomal dominant
Unknown
Table 1: Causative genes of CPVT.
RYR2, cardiac ryanodine receptor Ca2+ release channel
RYR2 is the first gene associated with the autosomal dominant form of CPVT [9]. Ryanodine channels open in response to Ca2+ entry from the outside of cardiomyocytes through L–type Ca2+ channel and release Ca2+ from the sarcoplasmic reticulum into the cytosol (Figure) [10]. Mutations in RYR2 are identified in ˜60% of patients with CPVT, and the vast majority of the genotype–positive CPVT patients have RYR2 mutations [4]. RyR2 channels open in response to Ca2+ entry from the outside of cardiomyocytes through L–type Ca2+ channel and release Ca2+ from the sarcoplasmic reticulum into thecytosol (Figure 1) [10]. There have been three proposed mechanisms underlying leaky RyR2 channels that cause spontaneous Ca2+ release leading to CPVT as a result of RYR2 mutations: 1) enhanced basalactivity of RyR2 channels [11–13]. , 2) disruption of ryanodine channel–stabilizing protein FKBP12.6 (or calstabin2) binding, [14]and 3) defective inter–domain folding to ryanodine receptor [15,16].
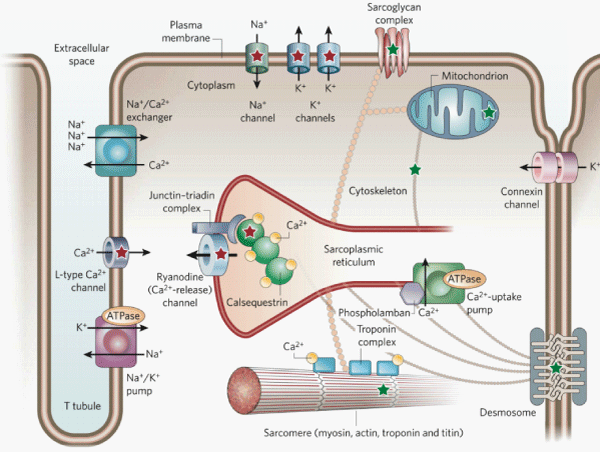
Figure 1: Protein complexes, cardiomyocyte architecture, and intracellular organelles involved in excitation–contraction coupling. Entry of Ca2+ into the cell triggers the release of Ca2+ from the sarcoplasmic reticulum through the ryanodine channel. Ca2+ then binds to the troponin complex and activates the contractile apparatus (the sarcomere, bottom). Relaxation occurs on removal of Ca2+ from the cytosol via Ca2+ uptake into the sarcoplasmic reticulum where Ca2+ binds to calsequestrin and via Ca2+ transport out the cell by the Na+ /Ca2+ exchanger. Modified from reference 5.
CASQ2, cardiac calsequestrin
CASQ2 is associated with the autosomal recessive form of CPVT, and mutations in CASQ2 are identified only in 1% to 2% of patients with CPVT patients [17]. Calsequestrin is the major Ca2+ storage protein in the sarcoplasmic reticulum and plays an essential role in the regulation of Ca2+ storage and release required for excitationcontraction coupling [18]. Calsequestrin forms a complex with the ryanodine channels and the junctional sarcoplasmic reticulum membrane proteins triadin 1 and junctin (Figure 1). Calsequestrin is also important as a regulator of ryanodine channels through the interaction with triadin and junctin, independent of its role for global Ca2+ buffering in the sarcoplasmic reticulum. Because CASQ2 is linked to the recessive form of CPVT, Casq2 null mice can be used in order to understand the mechanism underlying this form of CPVT. Casq2 null mice show increase in Ca2+ leak from sarcoplasmic reticulum, premature spontaneous Ca2+ releases, and exercise– and catecholamine–induced VT [19]. The mechanisms by which mutations in CASQ2 cause CPVT are various, including decreases Ca2+–storing capacity in the sarcoplasmic reticulum, altered Ca2+ sensitivity, reduced binding to triadin–1 and junctin, disrupted interaction with RyR2 channels, and decreased expression levels of calsequestrin [20–24].
TRDN, triadin
Triadin binds to both ryanodine channel and calsequestrin, and is important for anchoring calsequestrin at the junction in specialized areas where sarcoplasmic reticulum forms junctions with the sarcolemma. Triadin knock–out mice exhibit Ca2+ overload, spontaneous Ca2+ releases from sarcoplasmic reticulum, and CPVT [25]. Mutations in TRDN with a recessive mode of transmission have recently been identified in 2 of 97 families [26]. Two mutations are nonsense ones that result in complete absent of triadin and thus can cause CPVT by same mechanism to triadin knock–out mice. Another mutation in TRDN results in intracellular retention and lack ofmutant triadin expression.
KCNJ2, inward rectifier K+ channel Kir2.1
KNCJ2 is a causative gene of Andersen–Tawil syndrome or long QT syndrome 7. However, some patients carrying a mutation in KCNJ2 do not have Andersen–Tawil syndrome phenotypes including periodic paralysis nor dysmorphic features, but have CPVT [27]. Furthermore, a heterozygous mutation in KCNJ2 has been identified in 1 of 50 proband with CPVT [28]. Interestingly, electrocardiograms show abnormal U wave in KCNJ2–associated CPVT, similar to Andersen–Tawil syndrome, although electrocardiograms at rest are completely normal in other genetic CPVT forms [27,28]. Mutations in KCNJ2 result in decreased inward rectifier K+ currents, but theprecise mechanism by which KCNJ2 mutations cause CPVT is unknown [27].
CALM1, calmodulin
Calmodulin is a ubiquitous, multifunctional calcium signaling protein, which is essential for myriad intracellular signaling. Calmodulin regulates activities of various ion channels and has a critical role for cardiac electrophysiology. Mutations in calmodulin genes have recently been associated with long QT syndrome, idiopathic ventricular fibrillation, and CPVT [29–31]. Mutant calmodulins associated with CPVT show enhanced binding to ryanodine channels and result in increased activity of ryanodine channel and high frequency of Ca2+ waves, possibly leading to CPVT, while mutant calmodulins associated with long QT syndrome do notshow these abnormalities [32].
Mechanisms of bidirectional VT
Bidirectional VT is one of the unique characteristics in CPVT, and optical mappings in hearts of mice heterozygously carrying a Ryr2 mutation showed the possible underlying mechanism of bidirectional VT [33]. There are alternating foci between the right and left ventricles during bidirectional VT and the foci are located at the site of insertion of the major Purkinje system branches. Evidence that the susceptibility to DADs and triggered activity are high in Purkinje cells in comparison to ventricular myocytes supports the arrhythmogenic role of Purkinje systems in CPVT [34]. Furthermore, the initial beats of polymorphic VT originate from the free–running Purkinje fibers in the mouse model, [33] and catheter ablation for premature beats triggering polymorphic VT has been effective in patients with CPVT [35].
References
- Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995; 91: 1512-1519.
- Hayashi M, Denjoy I, Extramiana F, Maltret A, Buisson NR, Lupoglazoff JM, et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation. 2009; 119: 2426-2434.
- Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001; 103: 485-490.
- Cerrone M, Napolitano C, Priori SG. Catecholaminergic polymorphic ventricular tachycardia: A paradigm to understand mechanisms of arrhythmias associated to impaired ca(2+) regulation. Heart Rhythm. 2009; 6: 1652-1659.
- Watanabe H, Knollmann BC. Mechanism underlying catecholaminergic polymorphic ventricular tachycardia and approaches to therapy. J Electrocardiol. 2011; 44: 650-655.
- Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AA, Knollmann BC. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009; 15: 380-383.
- Watanabe H, van der Werf C, Roses-Noguer F, Adler A, Sumitomo N, Veltmann C, et al. Effects of lecainide on exercise-induced ventricular arrhythmias and recurrences in genotype-negative patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2013; 10: 542-547.
- van der Werf C, Kannankeril PJ, Sacher F, Krahn AD, Viskin S, Leenhardt A, et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol. 2011; 57: 2244-2254.
- Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001; 103: 196-200.
- Blayney LM, Lai FA. Ryanodine receptor-mediated arrhythmias and sudden cardiac death. Pharmacol Ther. 2009; 123: 151-177.
- Jiang D, Xiao B, Zhang L, Chen SR. Enhanced basal activity of a cardiac Ca2+ release channel (ryanodine receptor) mutant associated with ventricular tachycardia and sudden death. Circ Res. 2002; 91: 218-225.
- Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, et al. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc Natl Acad Sci U S A. 2004; 101: 13062-13067.
- Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, et al. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005; 97: 1173-1181.
- Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, et al. FKBP12.6 deiciency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003; 113: 829-840.
- Ikemoto N, Yamamoto T. Regulation of calcium release by interdomain interaction within ryanodine receptors. Front Biosci. 2002; 7: d671-683.
- Uchinoumi H, Yano M, Suetomi T, Ono M, Xu X, Tateishi H, et al. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ Res. 2010; 106: 1413-1424.
- Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, et al. A missense mutation in a highly conserved region of casq2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in bedouin families from israel. Am J Hum Genet. 2001; 69: 1378-1384.
- Knollmann BC. New roles of calsequestrin and triadin in cardiac muscle. J Physiol. 2009; 587: 3081-3087.
- Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006; 116: 2510-2520.
- di Barletta MR, Viatchenko-Karpinski S, Nori A, Memmi M, Terentyev D, Turcato F, et al. Clinical phenotype and functional characterization of casq2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2006; 114: 1012-1019.
- Valle G, Galla D, Nori A, Priori SG, Gyorke S, de Filippis V, et al. Catecholaminergic polymorphic ventricular tachycardia-related mutations R33Q and L167H alter calcium sensitivity of human cardiac calsequestrin. Biochem J. 2008; 413: 291-303.
- Houle TD, Ram ML, Cala SE. Calsequestrin mutant D307H exhibits depressed binding to its protein targets and a depressed response to calcium. Cardiovasc Res. 2004; 64: 227-233.
- Terentyev D, Nori A, Santoro M, Viatchenko-Karpinski S, Kubalova Z, Gyorke I, et al. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ Res. 2006; 98: 1151-1158.
- Katz G, Shainberg A, Hochhauser E, Kurtzwald-Josefson E, Issac A, El-Ani D, et al. The role of mutant protein level in autosomal recessive catecholamine dependent polymorphic ventricular tachycardia (cpvt2). Biochemical pharmacology. 2013; 86: 1576-1583.
- Chopra N, Yang T, Asghari P, Moore ED, Huke S, Akin B, et al. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation- contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci U S A. 2009; 106: 7636-7641.
- Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, Fauconnier J, Brocard J, Denjoy I, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Human molecular genetics. 2012; 21: 2759-2767.
- Kimura H, Zhou J, Kawamura M, Itoh H, Mizusawa Y, Ding WG, et al. Phenotype variability in patients carrying KCNJ2 mutations. Circ Cardiovasc Genet. 2012; 5: 344-353.
- Kawamura M, Ohno S, Naiki N, Nagaoka I, Dochi K, Wang Q, et al. Genetic background of catecholaminergic polymorphic ventricular tachycardia in japan. Circ J. 2013; 77: 1705-1713.
- Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013; 127: 1009-1017.
- Marsman RF1, Barc J2, Beekman L1, Alders M3, Dooijes D4, van den Wijngaard A5, et al. A Mutation in CALM1 Encoding Calmodulin in Familial Idiopathic Ventricular Fibrillation in Childhood and Adolescence. J Am Coll Cardiol. 2014; 63: 259-266.
- Nyegaard M, Overgaard MT, Søndergaard MT, Vranas M, Behr ER, Hildebrandt LL, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012; 91: 703-712.
- Hwang H, Nitu R, Yang Y, Johnson C, Faggioni M, Chazin W, et al. Calmodulin mutations in human genetic arrhythmia disorders exhibit divergent functional effects on ryanodine receptor ca release channels. Circulation. 2013; 128: A14157.
- Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O’Connell R, Berenfeld O, et al. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007; 101: 1039-1048.
- Kang G, Giovannone SF, Liu N, Liu FY, Zhang J, Priori SG, et al. Purkinje cells from RyR2 mutant mice are highly arrhythmogenic but responsive to targeted therapy. Circ Res. 2010; 107: 512-519.
- Kaneshiro T, Naruse Y, Nogami A, Tada H, Yoshida K, Sekiguchi Y, et al. Successful catheter ablation of bidirectional ventricular premature contractions triggering ventricular ibrillation in catecholaminergic polymorphic ventricular tachycardia with ryr2 mutation. Circ Arrhythm Electrophysiol. 2012; 5: e14-17.