
Department of Clinical Cardiology, Ospedale San Raffaele, Italy
*Corresponding author: Gabriele Fragasso, Department of Clinical Cardiology, Heart Failure Unit, Istituto Scientifico San Raffaele, Via Olgettina 60, 20132 Milano, Italy
Received: September 09, 2014; Accepted: November 11, 2014; Published: November 12, 2014
Citation: Loiacono F, Margonato A and Fragasso G. Modulation of Myocardial Metabolism by 3-Ketoacyl Coenzyme a Thiolase Inhibition and Other Agents in Heart Failure Treatment. Austin J Clin Cardiolog. 2014;1(5): 1031. ISSN 2381-9111
All cardiac syndromes may induce alterations of cardiac metabolism. Heart failure may itself promote metabolic changes such as insulin resistance, in part through neurohumoral activation, and determining an increased utilization of non-carbohydrate substrates for energy production. In fact, fasting blood ketone bodies as well as fat oxidation have been shown to be increased in patients with heart failure. The result is depletion of myocardial ATP, phosphocreatine and creatine kinase with decreased efficiency of mechanical work. A direct approach to manipulate cardiac energy metabolism consists in modifying substrate utilization by the failing heart. To date, the most effective metabolic treatments include several pharmacological agents, such as trimetazidine and perhexiline that directly inhibit fatty acid oxidation. These agents have been originally adopted to increase the ischemic threshold in patients with effort angina. However, the results of current research is supporting the concept that shifting the energy substrate preference away from fatty acid metabolism and toward glucose metabolism could be an effective adjunctive treatment in patients with heart failure, in terms of left ventricular function and glucose metabolism improvement. In fact, these agents have also been shown to improve overall glucose metabolism in diabetic patients with left ventricular dysfunction. Moreover, recent meta-analysis and a multicenter retrospective study have shown that additional use of trimetazidine in patients with heart failure, along with symptoms and cardiac function improvement also provides a significant protective effect on all-cause mortality, cardiovascular events and hospitalization due to cardiac causes. Nevertheless, the exact role of metabolic therapy in heart failure is yet to be established, and a large multicenter randomized trial is necessary.
In this paper, the recent literature on the beneficial therapeutic effects of modulation of cardiac metabolic substrates utilization in patients with heart failure is reviewed and discussed.
Keywords: Heart failure; Metabolic therapy; Trimetazidine; Free fatty acids inhibitors; Carnitine palmitoyl transferase I; Left ventricular function; Myocardial metabolism
ACE: Angiotensin Converting Enzyme; ATP: Adenosine Triphosphate; BNP: B-type Natriuretic Peptide; cGMP: Cyclic Guanosine Monophosphate; CoQ: Coenzyme Q; CPT: Carnitine Palmitoyl Transferase; ET-1: Endothelin-1; FFA: Free Fatty Acids; HF: Heart Failure; 3-KAT: 3-Ketoacyl Coenzyme A Thiolase; NO: Nitric Oxide; PCr: Phosphocreatine; XO: Xanthine Oxidase
Heart Failure (HF) has been traditionally treated through modification of hemodynamic alterations occurring in the failing heart, but administration and research on positive inotropes and drugs targeted at improving hemodynamics have yielded disappointing results. This is mainly due to the mechano-energetic uncoupling that characterize HF, i.e. an imbalance between left ventricular performance and myocardial energy consumption. Despite markedly impaired left ventricular work, the oxygen cost of contraction remains relatively unchanged, resulting in a decrease in the mechanical efficiency of contraction (Figure 1) [1]. On the contrary, the introduction of drugs intended to modulate neurohormonal activation consequent to HF, has been shown to slow disease progression. Despite these advancements, morbidity and mortality due to HF remain high, indicating that the development of adjunctive pharmacological agents and the identification of alternative therapeutic targets and strategies is needed. Recent studies have investigated the possibility of increasing cardiac performance without affecting oxygen consumption and hemodynamics, by agents aimed at enhancing myocardial energy efficiency.
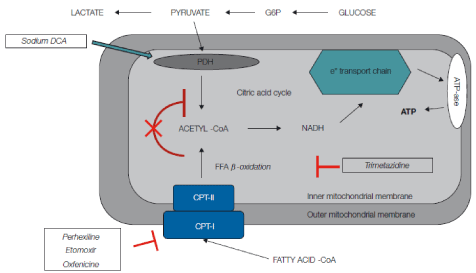
Most investigators have focused their efforts on agents that shift energy substrates utilization away from fatty acid metabolism and towards glucose metabolism, which is more efficient in terms of ATP production per mole of oxygen utilized. Carbohydrate metabolism may be directly increased by agents such as sodium dichloroacetate that stimulates pyruvate dehydrogenase activity by inhibiting pyruvate dehydrogenase kinase [2]. Stimulation of pyruvate dehydrogenase activity leads to enhanced glycolysis of glucose and utilization of lactate by the myocardium for aerobic respiration. Myocardial consumption of Free Fatty Acids (FFA) is simultaneously inhibited, with the overall effect of a change of substrate utilization from predominantly non-esterified fatty acids to glucose and lactate [3], finally resulting in improved left ventricular mechanical efficiency [4]. Alternatively, agents that directly inhibit fatty acid oxidation include 1) inhibitors of mitochondrial uptake of FFA via suppression of Carnitine Palmitoyl Transferase (CPT) I and II, 2) direct inhibitors of 3-Ketoacyl Coenzyme A Thiolase (3-KAT), the last enzyme involved in Β-oxidation (Figure 2). Of the latter pharmacological class, trimetazidine (1-[2,3,4-trimethoxybenzyl] piperazine dihydrochloride) is the most studied drug. Trimetazidine has been shown to affect myocardial substrate utilization by inhibiting oxidative phosphorylation and by shifting energy production from FFA to glucose oxidation [5]. Experimental evidence indicates that this effect is predominantly caused by a selective block of long chain 3-KAT [6]; however, this issue is still under debate [7,8]. These beneficial effects can be explained by the fact that by increasing utilization of glucose and lactate, which are more efficient fuels for aerobic respiration, the oxygen consumption efficiency of the myocardium can be improved by 16% to 26% [9]. In a very recent study Kuzmicic et al proposed a further possible mechanism of cellular protection for cardiomyocytes: trimetazidine protected cultured cardiomyocytes from palmitate-induced mitochondrial fission and dysfunction, increased intracellular lipid accumulation, and prevented palmitate-induced ceramide production, indicating that trimetazidine protects cardiomyocytes by changing intracellular lipid management and through modulation of the mitochondrial morphology and function [10]. These beneficial effects could be also effective in the skeletal muscles, as in vitro skeletal muscle models of atrophy indicate that trimetazidine triggers autophagy and counteracts stress-induced atrophy in skeletal muscle myotubes [11].
Fasting blood ketone bodies [12] as well as fat oxidation during exercise [13] have been shown to be increased in patients with HF. Insulin resistance has also been found associated with HF [14] and the consequent impaired suppression of lipolysis could determine the development of ketosis. Additionally, heart and arm skeletal muscle glucose uptakes are inversely related to serum Free Fatty Acid (FFA) levels [15] and increased FFA flux from adipose tissue to non-adipose tissue amplifies metabolic derangements that are characteristic of the insulin resistance syndrome [16]. New findings also suggest that raised FFA levels do not only impair glucose uptake in heart and skeletal muscle but also cause alterations in the metabolism of vascular endothelium leading to premature cardiovascular disease [17]. Therefore, FFA inhibitors could also play additional beneficial roles in terms of myocardial metabolism homeostasis.
Based on the hypothesis that FFA inhibitors could act as metabolic modulators in the protection of ischemic myocardium, Brottier and Colleagues assessed the value of long term treatment with trimetazidine in patients with severe ischemic cardiomyopathy, who were already receiving conventional therapy [18]. Twenty patients were randomized to either placebo or trimetazidine. All patients on trimetazidine, at 6 months follow-up, reported a clinically considerable improvement in symptoms and showed a higher ejection fraction compared to patients on placebo. The Authors concluded their study recommending the use of trimetazidine as a complementary therapeutic tool in patients with severe ischemic cardiomyopathy.
Subsequently, the effects of trimetazidine on dobutamine-induced left ventricular dysfunction in patients with angiographically proven coronary artery disease were assessed [19]. Patients were blindly and randomly assigned to a 15 day treatment period with either placebo or trimetazidine. They were then crossed over to the other regimen for 15 additional days. At the end of each treatment period, a stress echo with dobutamine was performed. Both in resting condition and at peak dobutamine infusion, wall motion score index was significantly lower on trimetazidine therapy than on placebo. Furthermore, trimetazidine induced an increase in dobutamine infusion time and an increase of the administered dobutamine dose to the development of ischemia. These results indicated that trimetazidine may not only protect from dobutamine-induced ischemic dysfunction, but could also improve resting regional left ventricular function, as shown by the significantly decreased peak and resting wall motion score index, during the active treatment period. A subsequent study confirmed these preliminary results [20].
The concept that 3-KAT inhibitors should be able to promote the utilization of glucose and non fatty substrates by the mitochondria led to focus attention on HF, where maintenance of metabolic efficiency is a crucial issue.
Initially, the effects of the addition of trimetazidine to standard treatment of diabetic patients with ischemic dilated cardiomyopathy on symptoms, exercise tolerance and left ventricular function, were assessed [21]. Thirteen such patients on conventional therapy were randomly allocated in a double blind fashion to either placebo or trimetazidine, each arm lasting 15 days and then again with placebo or trimetazidine for 2 additional 6 month periods. Both in the short and long terms, trimetazidine showed a significant beneficial effect on left ventricular function and control of symptoms, compared to placebo. The observed short-term trimetazidine benefit was maintained in the long-term and contrasts with the natural history of the disease, as shown by the mild but consistent decrease of EF when on placebo. These results paved the way to additional studies, that have invariably confirmed the positive effects of trimetazidine in patients with post-ischemic left ventricular dysfunction [22-25]. More specifically, Di Napoli et al [24] observed that the improvement of left ventricular function was also paralleled by a reduction of the inflammatory response in patients treated with trimetazidine. Finally, a more recent study from the same Authors has shown that long-term trimetazidine significantly reduces all-cause mortality and HF hospitalization in patients with ischemic cardiomyopathy [26]. All these studies were conducted on patients already on conventional therapy which included Angiotensin-Converting Enzyme (ACE) inhibitors (sometimes substituted by angiotensin receptor antagonists), beta-blockers and, in a more limited amount of cases, spironolactone or other anti-aldosterone drugs.
The beneficial effect of trimetazidine on left ventricular function, has been attributed to preservation of Phosphocreatine (PCr) and Adenosintriphosphate (ATP) intracellular levels [27]. Previous clinical studies using phosphorus-31 magnetic resonance spectroscopy to measure PCr/ATP ratios in human myocardium have shown that this ratio is reduced in failing human myocardium [28]. The PCr/ATP ratio is a measure of myocardial energetics and its reduction may depend on imbalance of myocardial oxygen supply and demand [29] and reduction of the total creatine pool, a phenomenon known to occur in HF [30]. In a recent study performed in patients with HF of different ethiologies on full standard medical therapy, it has been observed that the trimetazidine-induced improvement of functional class and left ventricular function is associated to an improvement of PCr/ATP ratio, supporting the hypothesis that trimetazidine probably preserves myocardial high energy phosphate intracellular levels [31]. These results appear particularly interesting, especially in view of previous evidence indicating the PCr/ATP ratio as a significant predictor of mortality [32].
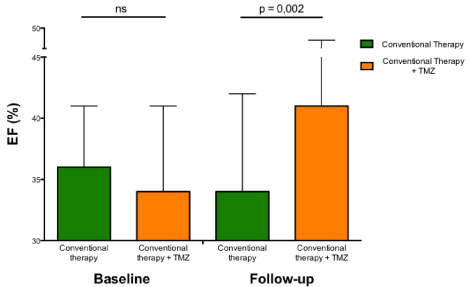
Based on the results of this pilot study, it has also been tested whether trimetazidine added to usual treatment, could also be beneficial in a more consistent group of patients with systolic-dysfunction HF of different etiologies [33]. Compared to patients on conventional therapy alone, those on trimetazidine improved functional class, exercise tolerance, quality of life and left ventricular function (Figure 3) and used less diuretics and less digoxin. Plasma B-type Natriuretic Peptide (BNP) level was also significantly reduced in patients on trimetazidine, compared to conventional therapy alone. These beneficial effects on left ventricular function could explain the subsequent observation of the anti-arrhythmic effect of trimetazidine in patients with post-ischemic HF [34].
A recent study has evidenced that energy deficiency in HF might result from increased uncoupling proteins (ie, less efficient ATP synthesis) and depleted glucose transporter protein (i.e., reduced glucose uptake) [35]. On this ground, the adoption of drug therapies such as 3-KAT inhibitors, aimed at interrupting the metabolic vicious circle in HF, has been advocated [36].
Finally, a recent international multicenter retrospective study on more than 600 patients demonstrated that patients on trimetazidine on top of conventional therapy showed a significant reduction of overall and cardiovascular morbidity and mortality at 5-year follow up compared to a control group on conventional therapy alone. This study, even if limited by its retrospective nature, confirmed in a large cohort of patients the potential usefulness of metabolic therapy with trimetazidine also in terms of prolonged overall and event-free survival [37].
Again, in all the reported clinical trials trimetazidine was tested on top of usual treatment for HF, which included neurohumoral counter-regulatory drugs such as ACE inhibitors and beta-blockers.
Overall, this data confirm that selective inhibition of 3-KAT represents a new therapeutic window in the treatment of patients with HF of different etiologies, and not only secondary to ischemic heart disease.
Modulation of abnormal glucose metabolism in heart failure: Regulation of glucose metabolism is an important target in the control of cardiovascular risk factors. Abnormalities of glucose homeostasis range from frank diabetes to a state of insulin resistance, a definition used to indicate the necessity to increase insulin levels in order to maintain normal glycemic levels. Recent studies have identified a direct relation between endothelial dysfunction and insulin resistance [38]. Endothelin-1 (ET-1) levels have been shown to significantly correlate with fasting insulin levels, systolic and diastolic blood pressure, visceral obesity and triglyceride levels, confirming a close relationship between insulin resistance and endothelial function [39]. When present, insulin resistance has been found to be operative in both cardiac and skeletal muscles [40]. Different degrees of endothelial dysfunction, often associated to a state of insulin resistance, have been evidenced in most cardiovascular diseases such as hypertension [41], coronary artery disease [42], microvascular angina [43] and HF [14]. On the other hand, insulin resistance is a pathological condition that is rarely diagnosed as a distinct entity. Our group has shown that more than 50% of patients submitted to coronary stenting for ischemic heart disease and with normal baseline blood glucose levels, present abnormal hyperglicemia after an oral glucose tolerance test, and that these abnormalities are associated to a higher probability of restenosis [44]. These results are supported by previous studies showing that impaired glucose tolerance not only runs the risk of developing overt diabetes and its associated microvascular complications but also has an increased risk of cardiovascular morbidity and mortality compared with healthy glucose-tolerant patients [45]. Therefore, early detection of impaired glucose tolerance would permit initiation of secondary preventive treatment measures in such patients.
Another possibility is to directly induce muscles to reduce FFA utilization in favor of glucose oxidation. In this context, the use of a partial fatty acid inhibitor could play a very specific role. In fact, as previously outlined, most cardiac diseases are associated to combined insulin resistance and endothelial dysfunction. In these contexts, improving the cardiac metabolic milieau by partially inhibiting FFA utilization could be particularly effective.
By keeping in mind the concept that 3-KAT inhibitors should, therefore, be able to promote the utilization of glucose and non fatty substrates by the mitochondria, attention has been focused on this specific issue. In fact, apart from improving left ventricular function in cardiac patients, it has been recently shown that trimetazidine could also improve overall glucose metabolism in the same patients, indicating an attractive ancillary pharmacological property of this class of drugs [21]. In fact, the known insulin resistant state in most cardiac patients is certainly aggravated in those patients with overt diabetes. This is particularly relevant in patients with both diabetes and left ventricular dysfunction. In this context, the availability of glucose and the ability of cardiomyocytes and skeletal muscle to metabolize glucose are grossly reduced. Indeed, since a major factor in the development and progression of HF is already a reduced availability of ATP, glucose metabolism alterations could further impair the efficiency of cardiomyocytes to produce energy. By inhibiting fatty acid oxidation, trimetazidine stimulates total glucose utilization, including both glycolysis and glucose oxidation. The effects of trimetazidine on glucose metabolism could therefore be dependent by a) improved cardiac efficiency; b) improved peripheral glucose extraction and utilization. Finally, considering the known relation between ET-1 concentration and glucose metabolism abnormalities [38,39] the observed beneficial effects of trimetazidine on glucose metabolism could also be partly ascribed to the positive effect of the drug on ET-1 levels reduction [21].
Animal studies have also suggested that trimetazidine improves blood glucose utilization in rats with fasting hyperglycemia [46] and more recently, prevents obesity-induced cardiomyopathy [47]. On this ground, both forearm glucose and lipid metabolism and forearm release of endothelial vasodilator and vasoconstrictor factors have been evaluated during a prolonged inhibition of β-oxidation by trimetazidine in patients with post-ischemic left ventricular dysfunction. Trimetazidine increased both insulin induced-forearm glucose oxidation and forearm cyclic-guanosine monophosphate release, while forearm ET-1 release was decreased [48]. Although these findings need further confirmation, the peripheral effects of trimetazidine add a new therapeutic window in the treatment of patients with ischemic heart disease and type 2 diabetes.
Finally, insulin sensitization with metformin in diabetic patients with HF, has been shown to reduce all-cause mortality [49,50]. Nevertheless, due to the perceived risk of acidosis, metformin has been previously formally contraindicated in HF. At present, metformin is being seriously reconsidered as a useful antidiabetic agent in HF [51].
An exhaustive paper has reviewed the basic science evidence, animal experiments, and human clinical data supporting the existence of an “insulin-resistant cardiomyopathy” and, partly based on the above discussed literature, proposed specific potential metabolic approaches [52].
Metabolic approach to endothelial dysfunction: Endothelial dysfunction, a critical component in the progression of HF, may result from increased oxidative stress, secondary to activation of the adrenergic and the renin-angiotensin systems and to the production of inflammatory cytokines, which in turn contribute to endothelial dysfunction [53]. In HF, the role of reduced bioavailability of Nitric Oxide (NO) is still under debate [54,55], while increased ET-1 levels are a mainstay [56]. It has been recently observed that trimetazidine could reduce endothelin release in cardiac patients [21,57]. Growth factors, vasoactive substances and mechanical stress are involved in the ET-1 increase in HF patients. Despite the known adaptative aspect of supporting contractility of the failing heart, persistent increases in cardiac ET-1 expression in the failing heart have a pathophysiological maladaptive aspect and are associated with the severity of myocardial dysfunction [58]. Trimetazidine-induced reduction of intracellular acidosis in ischemic myocardium could not only influence myocardial but also endothelial membranes [59]. By decreasing endothelial damage, trimetazidine could inhibit ET-1 release that, in turn, could decrease myocardial damage. A second hypothesis is that, by just decreasing the effects of chronic myocardial ischemia, trimetazidine could inhibit ET-1 release. Therefore, the observed decrease in ET-1 release with trimetazidine, could likely be linked to trimetazidine-induced reduction of myocardial ischemia. Finally, keeping in mind the close relation between endothelium and insulin sensitivity, the observed effects of trimetazidine on endothelial function could also explain the beneficial action of trimetazidine on glucose metabolism.
In the same context, the potential beneficial effect of six weeks oral L-arginine supplementation on endurance exercise, an important determinant of daily-life activity in patients with chronic stable HF, has been assessed [60]. L-Arginine is the precursor of endogenous Nitric Oxide (NO), which is potent vasodilator acting via the intracellular second-messenger cGMP. In healthy humans, L-arginine induces peripheral vasodilation and inhibits platelet aggregation due to an increased NO production. The results of this study show that arginine enhances endurance exercise tolerance, reducing both heart rate and circulating lactates, suggesting that chronic arginine administration might be useful as a therapeutic adjuvant in order to improve the patient’s physical fitness.
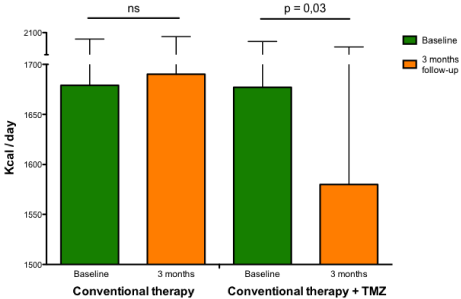
A higher resting metabolic rate has been observed in patients with HF [61-63] and this factor probably contributes to progressive worsening of the disease. Rate of energy expenditure is related to increased serum FFA oxidation and both energy expenditure and serum FFA oxidation are inversely correlated with left ventricular ejection fraction and positively correlated with growth hormone concentrations, epinephrine and norepinephrine [64]. Norepinephrine increases whole body oxygen consumption, circulating FFA concentrations, and FFA oxidation [65]. These changes have been attributed to stimulation of hormone-sensitive lipase in adipose tissue, and to stimulation of oxygen consumption independent of lipolysis by norepinephrine [66]. This data, together with close correlations between plasma norepinephrine concentrations, energy expenditure at rest and FFA oxidation, make increased sympathetic activity the most likely explanation for alterations in fuel homeostasis in patients with HF [66]. Therefore, intervention strategies aimed at optimizing global and cardiac metabolism, could be useful for interrupting the vicious circle of reduced function at greater metabolic expenses in different cardiac conditions [67]. In a recent study, it has been shown that 3 months treatment with trimetazidine added to usual treatment consistently reduces whole body resting energy expenditure (Figure 4) along with improved functional class, quality of life and left ventricular function in patients with systolic HF, regardless of its etiology and diabetic status [68]. The observation that the beneficial effect of trimetazidine on left ventricular function is also paralleled by a reduction of whole body rate of energy expenditure when compared to patients on conventional treatment underlies the possibility that the effect of trimetazidine may be mediated through a reduction of metabolic demand at the level of the peripheral tissues and, in turn, in some sort of central (cardiac) relief. Therefore, reduction of whole body energy demand could be one of the principal mechanisms by which trimetazidine could improve symptoms and left ventricular function in patients with HF.
A systematic literature search was conducted by Gao and Colleagues to identify randomised controlled trials of trimetazidine for HF [69]. They considered reports of trials comparing trimetazidine with placebo control for chronic HF in adults, with outcomes including all-cause mortality, hospitalisation, cardiovascular events, changes in cardiac function parameters and exercise capacity. The results of the search identified 17 trials with data for 955 patients. Trimetazidine therapy was associated with a significant improvement in left ventricular ejection fraction in patients with both ischemic (7.37%; 95% CI 6.05 to 8.70; p<0.01) and non-ischemic HF (8.72%; 95% CI 5.51 to 11.92; p<0.01). With trimetazidine therapy, New York Heart Association classification was also improved (p<0.01), as was exercise duration (p<0.01). More importantly, trimetazidine had a significant protective effect for all-cause mortality (RR 0.29; 95% CI 0.17 to 0.49; p<0.00001) and cardiovascular events and hospitalisation (RR 0.42; 95% CI 0.30 to 0.58; p<0.00001).
Finally, a recent meta-analysis has confirmed that additional use of trimetazidine in heart failure patients may decrease hospitalization for cardiac causes, improve clinical symptoms and cardiac function, and simultaneously ameliorate left ventricular remodelling [70].
Table 1 summarizes results on left ventricular function and other soft and hard endpoints from principal clinical trials of trimetazidine in HF.
Altogether, this data confirm that trimetazidine might be an effective strategy for treating HF and that a large multicentre randomized controlled trial should be performed, in order to clarify its exact therapeutic role in this setting.
Trimetazidine is registered as an anti-anginal drug. Its use in heart failure is at present an off-label indication. Trimetazidine is generally a safe and well tolerated drug. Recently, the European Medicines Agency (EMA) published a recommendation statement following a review of single reports series of movement disorders-related adverse events occurrence [71]. As a consequence, it is now suggested to discontinue trimetazidine in case of Parkinson symptoms, restless leg syndrome, tremors and gait instability occurrence. All these symptoms fully recovered after few months of discontinuation. Other contraindications only include severe renal failure and pregnancy. It has never been reported in any series an increased risk of mortality for patients undergoing trimetazidine, independently of underlying disease.
Trimetazidine has been commercialized since the 1970s, firstly in Europe and subsequently in many countries outside Europe. European countries where it is approved are Bulgaria, Cyprus, Czech Republic, Denmark, Estonia, France, Germany, Greece, Hungary, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Poland, Portugal, Romania, Slovakia, Slovenia and Spain. Many countries in Northern and Southern America, Asia and Africa have approved its use since decades.
Metabolic effects of beta-blockers in heart failure: Lowering raised plasma triglycerides and FFA levels could be the first therapeutic option to decrease the heart’s reliance on fatty acids and overcome the fatty acid inhibition of myocardial glucose utilization. Indeed beta-blockers, by reducing peripheral lypolysis, should reduce FFA availability. Interestingly enough, a recent study has shown that one of the main effects of the beta blocker carvedilol is the reduction of FFA utilization in favor of greater glucose utilization in patients with stable NYHA functional class III HF [72]. This change in myocardial energetics could provide a potential mechanism for the decreased myocardial oxygen consumption and improved energy efficiency seen with Β-adrenoreceptor blockade in the treatment of HF. The issue of whether non selective, compared to selective Β-adrenoreceptor blockers, are more efficient in shifting total body substrate utilization from lipid to glucose oxidation [73] is still controversial [74]. Nevertheless, a better metabolic attitude of the former could be one of the reasons of better survival rates observed with their use [75]. Additionally, central inhibition of sympathetic nervous activity with moxonidine in HF has been associated with increased mortality [76]. In fact, despite a significant reduction of cathecolamine spillover, moxonidine has been shown to increase FFA utilization and increase myocardial oxygen consumption [77]. This could be the reason for the failure of central sympathetic inhibition to prevent deaths in long term studies in patients with HF and also indicates that the predominant mechanism of action of beta-blockers is probably related to their peripheral antilypolytic action.
Other inhibitors of fatty acids oxidation: Etomoxir, perhexiline and oxfenicine are Carnitine Palmitoyl Transferase I (CPT-I) inhibitors. CPT-I is the key enzyme for mitochondrial FFA uptake; its inhibition, therefore, reduces FFA oxidation and their inhibitory effect on pyruvate dehydrogenase. As a consequence, glucose oxidation is increased [78,79]. Etomoxir, initially developed as an antidiabetic agent, has then been observed to improve left ventricular performance of pressure-overloaded rat heart [80]. These effects have been considered due to a selective modification of gene expression of hypertrophic cardiomyocytes [81]. Etomoxir could also increase phosphatase activation, have a direct effect on peroxisome proliferator activated receptor-alpha and up-regulate the expression of various enzymes involved in beta-oxidation [81]. The first clinical trial employing etomoxir in HF patients has shown a significant clinical and cardiac function improvement [82]. In experimental animal studies, etomoxir has also been shown to improve glucose metabolism [83]. However, the use of etomoxir may be limited by the observation that it may cause cardiac hypertrophy [84], oxidative stress [85] and increase liver transaminase levels [86].
Analogously to etomoxir, oxfenicine and perhexiline, originally classified as calcium antagonists, reduce cardiac utilization of long chain fatty acids by inhibiting CPT-I [87-90]. They have been initially developed as antianginal agents [90,91]. However, they have been recently employed in patients with HF. Metabolic modulation with perhexiline has been shown to improve O2 max, left ventricular ejection fraction, symptoms, resting and peak stress myocardial function, and skeletal muscle energetics [92]. Therefore, similarly to 3-KAT inhibitors, CPT-I inhibitors may represent a novel treatment in patients with HF with a good safety profile, provided that the dosage is adjusted according to plasma levels. In fact, perhexiline should be used with caution because of reports of hepatotoxicity and peripheral neuropathy [93,94].
Other drugs: L-carnitine is an essential cofactor of fatty acid metabolism, shuttling the end-products of peroxisomal fatty acid oxidation into the mitochondria and modulating the intramitochondrial acyl-coenzyme A/coenzyme A ratio. Although its main role is enhancement of FFA metabolism, experimental evidence also supports an enhancement of glucose metabolism. Several human and animal studies support a modest benefit in left ventricular energetics and function with L-carnitine administration [95-97]. Administration of the related propionyl-L-carnitine to the injured rat myocardium results in improved functional recovery and glucose use, supporting the theory that L-carnitine’s beneficial effects are due to its ability to increase glucose oxidation despite elevated FFA levels [95,97].
Ranolazine is a piperazine-derivate drug registered in Europe, Asia and USA for the treatment of chronic stable angina. Ranolazine exerts its anti-anginal effect mainly through the modulation of the late sodium current, thereby reducing the accumulation of intracellular Ca2+. Ranolazine is able to modulate other intracellular ionic currents thus also exerting an anti-arrhythmic effect. For the same reason, however, the drug has also a pro-arrhythmic activity; in fact one of the most dangerous side effects associated with the administration of ranolazine is the QT interval elongation. For this reason strict ECG monitoring is indicated in patients receiving ranolazine [98].
The anti-anginal efficacy and safety of ranolazine in diabetic and non-diabetic patients included in the Combination Assessment of Ranolazine In Stable Angina (CARISA) trial [99] were studied. Glycaemic control was also assessed in CARISA and its long-term open-label extension study. The anti-anginal efficacy and safety of ranolazine for angina were similar between diabetic and non-diabetic patients.
It has also been observed that ranolazine administration results in partial inhibition of free fatty acids oxidation, thus increasing the rate of glucose oxidation in the isolated rat heart, [100] even though the concentrations needed for metabolic action are greater than those achieved with normal therapeutic doses (100 mmol/l vs. 10 mmol/ l) [98]. Nevertheless, the MERLIN-TIMI 36 trial has demonstrated the efficacy of ranolazine in reducing one year mortality in patients with acute coronary syndrome and subsequent heart failure (BNP values rising > 80 pg/ml) [101]. Further analysis of the Merlin-TIMI 36 data has also shown that ranolazine improves glycemic control in patients with acute coronary syndrome and no ST elevation [102]. Additionally, ranolazine significantly improved glycaemic control in diabetic patients [103] confirming that, apart from their primary cardiac action, this class of drugs yields also this important ancillary effect on glucose metabolism. Future studies will clarify the potential role of ranolazine as a metabolic modulator in patients with heart failure and diabetes.
In a very recent study, spironolactone has been attributed with direct metabolic effects on myocardium apart from its specific anti-aldosterone activity. Twelve patients with nonischemic dilated cardiomyopathy underwent a ≥6 months spironolactone therapy added to a standard HF regimen; the Work Metabolic Index (WMI), an index of left ventricle mechanical efficiency, and kmono/RPP (rate-pressure product), an index of energy supply/demand, were assessed through [11C] acetate Positron Emission Tomography (PET), at baseline and after spironolactone. The WMI increased (P=0.001), as did kmono/RPP (P=0.003). These improvements were associated with reverse remodeling, increased left ventricle ejection fraction, and decreases in LV mass and systolic wall stress (all P<0.002). These effects cannot however be fully ascribed to a direct metabolic action of the drug. In fact, its unloading diuretic effect associated to antifibrotic properties, could well be the reasons of the observed improved myocardial metabolism [104].
Xantine oxidase inhibition could also play a role. The mechanisms responsible for mechanoenergetic uncoupling are unknown, although experimental evidence suggests that reactive oxygen species may play a major role. Markers of reactive oxygen species accumulate in HF patients [105-106], indicating that HF is a state of oxidative stress [107]. Although there are several potential sources of reactive oxygen species, increased levels of uric acid in the serum of HF patients suggest that Xanthine Oxidase (XO) activity contributes [108]. XO inhibition with allopurinol improves myocardial efficiency [109] and enhances the contractile response of failing myocardium to dobutamine and to exercise in animal [110] and human [111] models of HF. Because XO inhibitors have a well-established safety profile and are used widely for the treatment of gout, they have the potential to be rapidly tested as a novel metabolic therapy for the treatment of human HF.
Mitochondrial metabolic oxidants: coenzyme Q and lipoic acid: Coenzyme Q (CoQ) does not serve as a coenzyme in mitochondrial oxidation, despite its name. Lipoic acid is a cofactor of the pyruvate dehydrogenase enzyme system. The presence of these molecules indicates that mitochondria can defend themselves against harmful effects of the oxygen atmosphere [112-114]. Coenzyme Q, also called ubiquinone or ubidecarenone, in its reduced form is an excellent antioxidant. The name “ubiquinone” also indicates that it can occur in various places. The endogenous coenzyme Q can capture perferril, carbon-centred lipids, lipid-peroxyl and alkoxyl radicals.
Coenzyme Q has been shown to be beneficial due to its inhibitory effect on lipid peroxidation and on the oxidation of endogenous coenzyme Q-9 as well as by improving mitochondrial respiration [114,115]. Recently, oral CoQ10 has been shown to improve functional capacity, endothelial function, and left ventricular contractility in patients with HF without any side effects [116]. Despite anecdotical evidence to support the therapeutic value of CoQ10 as an adjunct to standard medical therapy in congestive HF, much further research is required, especially to assess functional outcomes in patients with congestive heart failure [117,118].
Lipoic acid is synthesized both in the animal and in the human body [119]. This fatty acid with eight carbon atoms containing disulphide groups at the sixth and eighth carbon atoms closed in a pentamerous ring is essential for the function of mitochondrial pyruvate dehydrogenase. Both lipoic acid and its reduced form are excellent metabolic antioxidants, as they take part in the antioxidant redox cycle of the organism. Despite the potential beneficial effects of lipoic acid on myocardial metabolism, there are not yet studies showing its beneficial effects in heart disease.
Other micronutrients: Apart from defects in substrate metabolism and cardiac energy and substrate utilization, HF is often accompanied by a deficiency in key micronutrients required for unimpeded energy transfer. Correcting these deficits has been proposed as a method to limit or even reverse the progressive dysfunction in HF. A recent review has summarized the existing literature with respect to supplementation trials of key micronutrients involved in cardiac metabolism [120]. Although some of the results are promising, none are conclusive. There is a need for a prospective trial to examine the effects of micronutrient supplementation on morbidity and mortality in patients with HF.
Metabolic therapy could have an important role in the therapeutic strategy of patients with HF. In several small studies, shifting the energy substrate preference away from FFA metabolism and toward glucose metabolism has been shown to be an effective adjunctive treatment in patients with HF, in terms of left ventricular metabolism and function improvement. At present trimetazidine, a partial FFA oxidation inhibitor, appears as the most promising agent for the metabolic approach to HF. All principal clinical trials were conducted with addition of trimetazidine on top of conventional therapy, which included neuro-hormonal counter-regulatory drugs. This provides evidence that metabolic therapy efficiently works in cooperation with established therapeutic strategies. Although highly suggestive, whether the observed benefits would definitely translate into improved survival should be ascertained by a multicenter trial [121-123]. Time has come to test this huge potential therapeutic advancement in HF syndromes, which still suffer very high morbility and mortality rates.
Additionally, most cardiac diseases are associated to abnormalities of glucose homeostasis, which definitely contribute to the progression of the primary disease. If not adequately treated, in most cardiac patients glucose metabolism abnormalities will heavily contribute to the occurrence of complications, of whom severe left ventricular dysfunction is at present one of the most frequent and insidious. Apart from a meticulous metabolic control of frank diabetes, special attention should be also paid to insulin resistance, a condition that is generally under-diagnosed as a distinct clinical entity. The observed combined beneficial effects of FFA inhibitors on left ventricular function and glucose metabolism, represent an additional advantage of these drugs, especially in those cardiac patients in whom myocardial and glucose metabolism abnormalities coexist.
Overall, currently available data state that although the sub-population of HF patients with abnormal glucose metabolism and with ischemic heart disease or both may take the major advantage from trimetazidine use, actually a great clinical benefit has been observed in HF independently from comorbidities or aetiology and therefore it could be administered to all patients as an adjunctive drug on top of optimized therapy according to current guidelines. Nevertheless, we recognize the higher amount of evidence regarding these two subtypes of HF patients and therefore we feel that especially in these cases trimetazidine should be considered as a first-line additional medication.
Results from principal clinical trials of trimetazidine in systolic heart failure patients.
Trial |
Study design |
Number of patients |
Follow up |
Mean EF improvement |
Other endpoints |
Brottier et al, [18] |
Vs placebo |
20 |
6 months |
9.3% (p< 0.018) |
Improvement of dyspnea |
Fragasso et al, [21] |
Vs placebo |
16 |
a)15 days b)6 months |
a)5.9% (p<0.001) b)8.5% (p<0.001) |
Improvement of left ventricle end-sysytolic and end-dyastolic diameters and volumes |
Rosano et al, [22] |
Vs placebo |
32 |
6 months |
5.4% (p<0.05) |
Improvement of end-diastolic diameters, wall motion score index and E/A wave ratio |
Vitale et al, [23] |
Vs placebo |
47 |
6 months |
7.4% (p<0.0001) |
Improvement of left ventricle end-sysytolic and end-dyastolic diameters and volumes, wall motion score index, NYHA class and quality of life |
Di Napoli et al, [24] |
Vs conventional therapy alone |
61 |
a) 6 months b)12 months c)18 months |
a)2% (p<0.001) b)10% (p<0.001) c)11% (p<0.001) |
Improvement of NYHA class, end-sysytolic and end-dyastolic volumes |
Fragasso et al, [31] |
Vs placebo |
12 |
3 months |
5% (p=0.003) |
Improvement of cardiac PCr/ATP ratio, NYHA class and metabolic equivalent system |
Fragasso et al, [33] |
Vs conventional therapy alone |
55 |
13 +/- 3 months |
7% (p = 0.002) |
Improvement of NYHA class and end-systolic volume |
Sisakian et al, [25] |
Vs conventional therapy alone |
82 |
3 months |
3.5% (p=0.05) |
Improvement of tolerance to physical activity on 6 minutes walking test |
Fragasso et al, [37] |
Retrospective, vs conventional therapy alone |
669 |
5 years
|
n.a. |
Improvement of global survival, survival for cardiovascular death, hospitalization-free survival and reduction of hospitalization for cardiovascular causes |
Gao et al, [69] |
Meta-analyses |
955 |
- |
- 7.37% (p<0.01) (ischemic HF) - 8.72% (p<0.01) (non-ischemic HF) |
Reduction of all-cause mortality, cardiovascular events and hospitalizations; improvement of exercise duration and end-systolic volume |
Zhang et al, [70] |
Meta-analyses |
884 |
- |
6.46% (p<0.0001) |
Reduction of hospitalization for cardiac causes; improvement of total exercise time, NYHA class, B-type natriuretic peptide levels and end-systolic and end-diastolic diameter |
Abbreviation: TMZ: ATP: Adenosine Triphosphate; HF: Heart Failure; NYHA: New York Heart Association; PCr: Phosphocreatine
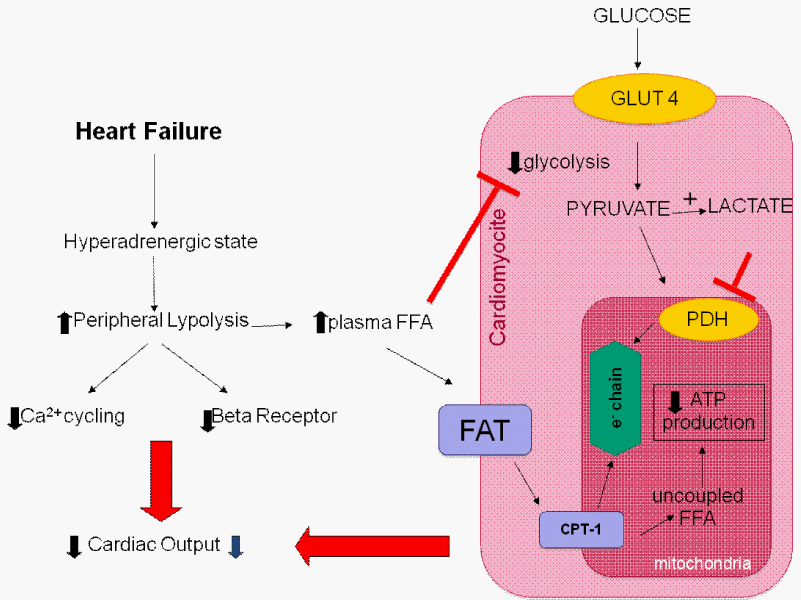
FFAs inhibit glycolysis and glucose uptake by the heart. Plasma FFA taken up by the heart is activated and transported by Carnitine Palmitoyltransferase-1 (CPT-1) into the mitochondria to uncouple respiration with oxygen wastage. Additionally, hyperadrenergic state downregulates beta-adrenergic receptors.
Abbreviations: Acetyl-CoA: Acetyl Coenzyme A; ATP: Adenosine Triphosphate; FFA: Free Fatty Acids; PDH: Pyruvate Dehydrogenase
Effects of metabolic drugs at mitochondria level. Carbohydrate metabolism may be directly increased by agents such as sodium dichloroacetate, which stimulates Pyruvate Dehydrogenase (PDH) activity by inhibiting pyruvate dehydrogenase kinase. Stimulation of PDH activity leads to enhanced glycolysis and utilization of lactate by the myocardium for aerobic respiration. Myocardial consumption of Free Fatty Acids (FFA) is simultaneously inhibited, with the overall effect of a change of substrate utilization from predominantly FFA to glucose and lactate. Perhexilline, oxfenicine, and etomoxir prevent the uptake of FFA by inhibiting carnitine palmitoyltransferase I, which is a key mitochondrial enzyme involved in this process. Trimetazidine inhibits β-oxidation of FFA. These actions shift myocardial substrate use from FFA to glucose, which is more efficient in terms of energy production, leading to an oxigen-sparing effect.
Abbreviations: CoA: Coenzyme A; CPT: Carnitine Palmitoyltransferase; DCA: Dichloroacetate; FA: Fatty Acid; G6P: Glucose-6-Phosphate.
Long-term effects of trimetazidine on ejection fraction in patients with heart failure of different etiologies. Histograms (mean±1 SD) show the significant beneficial long-term effects of trimetazidine compared with conventional therapy alone.
Abbreviations: EF: Ejection Fraction; ns: Not Significant; SD: Standard Deviation; TMZ: Trimetazidine.
Rate of energy expenditure in heart failure patients. Rate of energy expenditure (kcal/d) measured by indirect calorimetry at baseline and at 3-month follow-up in patients with heart failure receiving conventional therapy alone (left histograms) or conventional therapy plus trimetazidine (right histograms).
Abbreviations: ns: Not Significant; TMZ: Trimetazidine
Austin Publishing Group is an emerging open access publisher specialising in Science, Technology and Medicine is dedicated to serve the biomedical community through its initiatives. Austin Publishing Group is an academic publisher with 100+ peer reviewed open access journals in various subjects such as biomedical, Pharma, Life Sciences, Environmental, Engineering and Management. Austin Publishing Group publishes Open Access eBooks providing free access to vast scientific literature.