
Research Article
Austin J Comput Biol Bioinform. 2014;1(2): 6.
Graphics Card Processing: Acceleration of Multiple Sequence Alignment
Hanif MK* and Zimmermann KH
Institute of Computer Technology, Hamburg University of Technology, Germany
*Corresponding author: Muhammad Kashif Hanif, Institute of Computer Technology, Hamburg University of Technology, 21071 Hamburg, Germany
Received: September 16, 2014; Accepted: December 01, 2014; Published: December 05, 2014
Abstract
ClustalW is the most widely used heuristic method for multiple sequence alignment. It consists of three stages: distance matrix calculation, guide tree compilation, and greedy-fashion alignment. The high computational complexity demands methods to accelerate the algorithm. In this work, the efficient mapping of the progressive alignment stage onto graphics processing unit by using a combination of wavefront and matrix-matrix product techniques will be studied. The experimental results exhibit one order of magnitude speed-up over the serial version.
Keywords: Alignment; Progressive alignment; Graphics processor card; ClustalW; Performance
Introduction
Sequence alignment is the fundamental technique in molecular biology to compare sequences and to identify regions of similarity that are eventually consequences of structural, functional, or evolutionary relationships [1- 4]. Sequence alignment is performed for all kinds of organic molecules, like DNA, RNA, or protein sequences. Multiple sequence alignment is the technique to align three or more sequences simultaneously. The aligned sequences are obtained by inserting gaps and have equal length. However, multiple sequence alignment is very time-consuming. For instance, optimal dynamic programming methods require O(2knk) steps to simultaneously align k sequences of length O(n) [4]. A variety of heuristic methods have been developed to cope with multiple sequence alignment problems. The most widely accepted heuristic method for aligning multiple sequences is progressive alignment [5,6]. This method aligns more closely related sequences first and then gradually adds more divergent sequences [7]. The alignment accuracy can be improved by assessing the sequences according to their relatedness. A progressive alignment algorithm can handle a larger number of sequences in practical time scales. The most widely used progressive alignment programs are ClustalW [5, 8, 9], T-Coffee [6,10], MAFFT [11,12], and MUSCLE [13,14].
ClustalW is a typical progressive alignment algorithm making use of the policy "once a gap, always a gap", i.e., gaps introduced earlier in the alignment remain valid as new sequences are added [9,15]. It works in three stages (Figure 1). In the first stage, the distances between all pairs of sequences are calculated by pairwise sequence alignment. Pairwise sequence alignment can be calculated by the dynamic programming based method of Needleman-Wunsch [16] or one of its varieties like Smith-Waterman [17] or a fast heuristic method [9,18-20]. The scores of attained pairwise alignments are converted into distances which are input for the subsequent stage [9].
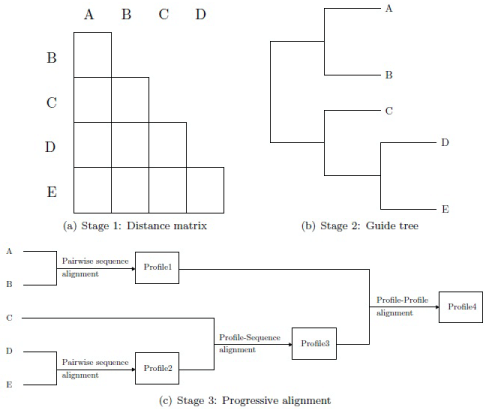
Figure 1: Stages of the ClustalW algorithm. The first stage computes the pairwise distances between the sequences. The guide tree is built in stage two using the distances. In stage three, the sequences are progressively aligned.
In the second stage, the distance matrix calculated in the first stage is used to build the guide tree which serves as a guide for the calculation of the overall multiple sequence alignment. This tree can be constructed by a heuristic phylogenetic method, like Neighbour joining [21] or unweighted pair group method with arithmetic mean (UPGMA) [22].
In the final stage, the sequences are progressively aligned u sing the guide tree. For this, the sequences correspond one-to-one with the leaves of the tree. Three cases can occur:
- An inner node (cherry) whose descendants are leaves is associated with the pairwise alignment of the sequences corresponding to these leaves.
- An inner node whose descendants are a leaf and an inner node is associated to the alignment given by the sequence and the multiple alignments. This can be achieved by profile-sequence alignment where the given multiple alignment is represented by a statistical representative called profile.
- An inner node whose descendants are two inner nodes is associated to the alignment given by the corresponding multiple alignments. This can be attained by profile-profile alignment where the given multiple alignments are represented by statistical representatives.
The root of the tree corresponds to the overall multiple sequence alignment. The basic algorithm uses one weight matrix and fixed gap opening and extension penalties.
This approach, however, is not suitable for more divergent sequences. In this case, sequence weights are calculated from the guide tree. Closely related sequences have lower weights while the divergent ones have higher weights. Moreover, different substitution matrices are used at different alignment stages. New penalties are calculated based on the length and similarity of sequences, weight matrix, and gap positions [4,9]. An example using the tat and vpu proteins from HIV 1 (Human Immunodeficiency Virus) is shown in Figure 2. The complexity of the ClustalW algorithm is shown in Table 1 where n is the number of sequences and l is the average sequence length [20].

Figure 2: ClustalW based sequence alignment between the tat and vpu proteins from HIV 1 calculated from EMBL-EBI using the BLOSUM substitution matrix. The gap opening and the gap extension penalties for pairwise alignments are 10 and 0.1, respectively, and the initial gap penalty and the gap extension penalty for multiple alignments are 25 and 0.2, respectively.
![]()
Stage
O(Time)
Distance matrix
O(n2l2)
Guide tree
O(n3)
Progressive alignment
O(nl2 + n2l)
Total
O(n2l2 + n3)
Table 1: Complexity of the ClustalW algorithm by stage [20].
Many efforts have been made to accelerate the performance of the ClustalW algorithm. ClustalW-MPI [23], Ebedes et al. [24], and pCLUSTAL [25] use MPI to parallelize ClustalW on a cluster. ClustalW-MPI parallelized all three stages and achieved approximately 4.3 speed-up using 16 processors. Ebedes et al. demonstrated a speed-up of 5.5 by parallelizing the stages one and three. Similarly, Tan et al. [26] use MPI/Open MP for symmetric multiprocessors to parallelize the stages one and three. Mikhailov et al. [27] show a 10-fold speed-up by parallelizing all three stages with OpenMP on a shared-memory SGI machine. Aung et al. [28] employed a Field-Programmable Gate Array (FPGA) for acceleration of stage one. Oliver et al. [29] mapped stage one on FPGA and attained a speed-up between 45 and 50. MT-ClustalW [30] utilized pthreads to parallelize all three stages. GPU-ClustalW [31] parallelized the first stage on a GPU with OpenGL to obtain approximately 7 speed-up. MSA-CUDA [19] exploited the parallel architecture of the GPU by implementing all three stages and achieved a maximum average speed-up of approximately 37 for a small number of long sequences. Bassoy et al. [32] formulated a matrix-matrix product algorithm by separating the profile-sequence alignment algorithm into a data dependent and a data independent part to attain an order of magnitude speed-up on a GPU. However, they have ignored the time taken by executing the data dependent part on the CPU which is the reason for their huge speed-up given. Recently, Hanif and Zimmermann [33] described parallel algorithms for profile-profile alignment using matrix-matrix product and the wavefront approach attaining a 20-fold average speed-up for the wavefront approach. The results have shown that the matrix-matrix product and the wavefront methods are the most promising for profile-sequence alignment and profile-profile alignment, respectively.
A Graphics Processing Unit (GPU) is a highly parallel many-core streaming architecture which can execute hundreds of threads in a concurrent manner. The data parallel architecture of a GPU is particularly suitable to perform computation intensive tasks. GPUs offer orders of magnitude more computation power than CPUs and are becoming increasingly popular for general purpose computations to attain high speed-ups. A large set of problems in molecular dynamics, physics simulations, and scientific computing [34] have been tackled by mapping them onto a GPU. NVIDIA has introduced a GPU programming model called Compute Unified Device Architecture (CUDA) which enables the programmer to write C-like functions called kernels with some extensions that leverages programmers to efficiently use the graphics API. Each kernel is executed by a batch of parallel threads. CUDA provides three key abstractions: a hierarchy of thread groups, shared memories, and barrier synchronization [34].
In this paper, a combination of matrix-matrix product and wavefront methods will be used to parallelize the progressive alignment stage of ClustalW. The paper is organized as follows: Section 2 provides a method to accelerate the progressive alignment stage of ClustalW and section 3 evaluates its performance and compares it to the CPU implementation.
Materials and Methods
The performance of the ClustalW algorithm can be improved using the parallel architecture of the GPU. This particularly holds for the third stage, the alignment of the sequences using a guide tree.
First, a matrix-matrix product based approach to profile-sequence alignment will be introduced [35]. The technique of Bassoy et al. [32] is similar, but required additional memory and clock cycles.
Given a multiple alignment of length m by its profile P on the alphabet Σ' = Σ'{-} and a sequence x of length n, the score between a column p of the profile and a character a∈ Σ' is [4]
Profile-sequence alignment algorithm can be converted into a matrix-matrix product by separating the data dependent and independent parts. First, the data independent part calculates three scalar products. The diagonal entries of the forward table are stored in m x n matrix D. Two vectors h and v of lengths m and n are used to store the vertical and horizontal entries of the forward table. Second, these values are used in the data dependent part for calculation of the forward table entries.
The profile column - p= (0,...,0,1)T represents a column consisting of blanks having relative frequency of blank 1. Take the extended alphabet Σ' = {a1,...,a1}, where a1 equals blank, and assign
wa=(σ(a,a1),...,σ(a,a1))T, a∈Σ'. (2)
The vectors h and v can be computed as
The matrix D of size m x n is calculated as
The calculations of h, v, D can be written into a matrix-vector product. For this, take the l x n matrix W
W = (wx1,...,wxn).
The values of the vectors h and v and the matrix D are determined as
v=PT .w-, (6)
To calculate first column of the forward table, take the lower triangular m x m matrix Bm
Similarly, the first row is calculated by having the n x n upper triangular matrix Bn
This gives the algorithm PROSEQALIGNMATVECPRODV2.
Algorithm 1: PROSEQALIGNMATVECPRODV2(x,P).
Require: sequence x = x1 . . . xn and profile P = p1 . . . pm
- S0,0 ← 0 {initialization}
- v ← PT • w-
- h ← -Tp • W
- s*,0 ← Bmv
- S0,* ← hBn
- for i ← 1 to m do {calculation}
- Di ← pTi •W
- end for
- for i ← 1 to m do {maximization}
- for j ← 1 to n do
- Si,j←max{Si-1,j+vi,Si,j-1+hj,Si-1,j-1+Dij}
- end for
- end for
- return S
The matrix D can be computed by matrix multiplication as
D = PT. W. (11)
This resultant algorithm is PROSEQALIGNMATPRODV2.
Five versions of profile-sequence alignment algorithm on GPU have been considered.
Algorithm 2: PROSEQALIGNMATPRODV2(x,P).
Require: sequence x = x1 . . . xn and profile P=p1...pm
- S0,0 ← 0 {initialization}
- v ← PT • w-
- h ← -Tp • W
- s*,0 ← Bmv
- S0,* ← hBn
- D ← PT.W
- for i ← 1 to m do {maximization}
- for j ← 1 to n do
- Si,j←max{Si-1,j+vi,Si,j-1+hj,Si-1,j-1+Dij}
- end for
- end for
- return S
- MatVecProd V1: Matrix-vector product implementation using cublasSgemv [32].
- MatVecProd V2: Matrix-vector product implementation using cublasSgemv.
- MatProd V1: Matrix-vector product implementation using cublasSgemm [32].
- MatProd V2: Matrix-vector product implementation using cublasSgemm.
- SMWavefront 256: Wavefront approach using shared memory having block size 256 [35].
For MatVecProd V1 and MatProd V1, results are taken up to sequence length 6,000. A sequence length of 10,000 requires approximately 1145 MB to store matrices which is well beyond the capacity of available global memory (1024 MB). A maximum speed-up factor of approximately 28 is attained when compared with optimized Intel CPU implementation (Figure 3). Parallel versions of profile-profile alignment are given in [33]. The results show that a mixture of wavefront and matrix-matrix product methods can be useful for the parallelization of the progressive alignment stage.
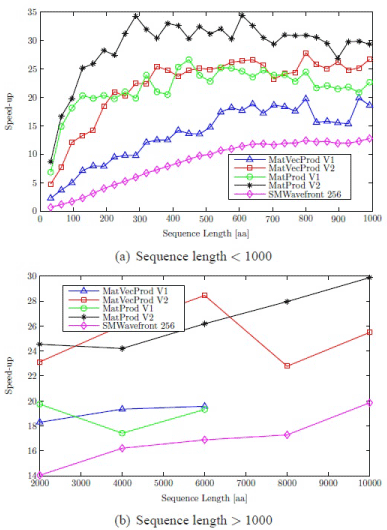
Figure 3: Speed-ups of profile-sequence alignment algorithms.
The approach adopted is similar to that in [19]. First, the intermediate nodes of the guide tree are labeled by post-order traversal. Two vectors are used to maintain the right child and left child of the nodes. One flag vector is required to keep track whether the node has been aligned. The flag for the leaf nodes is set to 1 when the alignment is not required. The left and right children indices are 0 for the leaf nodes.
The flag vector is checked to identify the nodes of the guide tree to be aligned. An alignment at an intermediate node can only be performed if the right and left children have been aligned. In first phase, the leaf nodes are aligned using pairwise sequence alignment since the left and right children are assumed to be aligned as indicated in the flag vector. In next phase, the flag vector is again checked to find potential candidates for subsequent alignment. This process continues until the overall multiple sequence alignment is obtained and the flag vector contains 1 for each node.
The frequency based profiles are constructed for the intermediate nodes. A profile and a sequence are aligned by the matrix-matrix product while two profiles are aligned using the wavefront method on the GPU. The traceback is performed on the CPU to find the alignment by adding gaps in the aligned sequences.
Results and Discussion
The ClustalW progressive alignment stage has been implemented on an Intel Core 2 Duo 6600 CPU (2.40 GHz) running openSUSE 11.4 linux distribution and CUDA version 4.0 on an NVIDIA GeForce GTX 560 Ti graphics card. The tests have been conducted using a serial gcc compiler (version 4.4.1) and an NVIDIA nvcc compiler. The performance of the parallel progressive alignment stage has been measured by the speed-up. The dataset for the implementation of the ClustalW algorithm is similar to that in [19]. The protein sequence dataset consists of the HIV dataset available at the NCBI database. The dataset has been divided into several subsets:
- 400 sequences of average length 856 and 1000 sequences of average length 858;
- 2000 sequences of average length 266 and 4000 sequences of average length 247;
- 4000 sequences of average length 57 and 8000 sequences of average length 73.
The execution times have been averaged over ten runs for each data subset. The times for memory allocation and data transfer to or from the GPU have been neglected. The input to the progressive alignment stage is a guide tree which has been generated by the ClustalW program from the EMBL-EBI website.
The speed-up for the progressive alignment stage is illustrated in Figure 4. Note that profile creation and traceback have been performed on the CPU and have not been neglected. The data subsets given by longer sequences have achieved a speed-up of one order of magnitude since the matrices to be multiplied utilize the hardware resources rather efficiently. The data subsets given by shorter sequences show a similar behavior due to the possibility to process multiple sequences in each pass. The computation-to-communication ratio of wavefront approach for sequences and profiles [33] of average length is low which impacts the speed-up exhibited by the data subsets of average length sequences. The maximum speed-up attained is much better than the speed-up of approximately 6 exhibited by progressive alignment stage of MSA-CUDA [19].
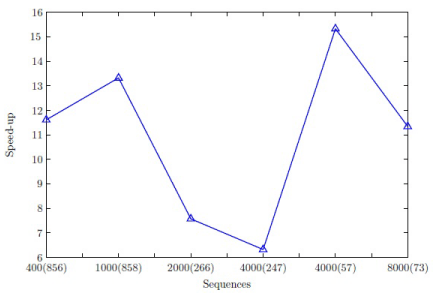
Figure 4: Speed-up for progressive alignment stage of the ClustalW.
Conclusion
This paper has provided a parallel algorithm for the progressive alignment stage of the ClustalW algorithm onto the GPU using a mixture of algorithms: matrix-matrix product for profile-sequence alignment and wavefront for profile-profile alignment. The results have shown a performance increase of more than one order of magnitude for several data sets considered.
Acknowledgement
The authors would like to thank Yongchao Liu for providing the dataset. The work of the first author has been sponsored by DAAD and Higher Education Commission of Pakistan.
References
- Durbin R, Eddy SR, Krogh A, Mitchison GJ. Biological sequence analysis: probabilistic models of proteins and nucleic acids. Cambridge University Press. 1998.
- Gusfield D. Algorithms on strings, trees, and sequences: computer science and computational biology. New York: Cambridge University Press. 1997.
- Waterman MS. Introduction to computational biology: maps, sequences, and genomes: interdisciplinary statistics. CRC Press. 1995.
- Zimmermann KH. An introduction to protein informatics. Boston: Kluwer Academic Publishers. 2003.
- Higgins DG, Sharp PM. CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene. 1988; 73: 237-244.
- Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. Journal of Molecular Biology. 2000; 302: 205-217.
- Feng DF, Doolittle RF. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J Mol Evol. 1987; 25: 351-360.
- Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003; 31: 3497-3500.
- Thompson J, Higgins D, Gibson T. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research. 1994; 22: 4673-4680.
- Magis C, Taly JF, Bussotti G, Chang JM, Di Tommaso P, Erb I, et al. T-Coffee: tree-based consistency objective function for alignment evaluation. Methods in Molecular Biology. 2014; 1079: 117-129.
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002; 30: 3059-3066.
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013; 30: 772-780.
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004; 32: 1792-1797.
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004; 5: 113.
- Higgins DG, Thompson JD, Gibson TJ. Using CLUSTAL for multiple sequence alignments. Methods Enzymol. 1996; 266: 383-402.
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970; 48: 443-453.
- Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981; 147: 195-197.
- Bashford D, Chothia C, Lesk AM. Determinants of a protein fold. Unique features of the globin amino acid sequences. J Mol Biol. 1987; 196: 199-216.
- Liu Y, Schmidt B, Maskell DL. MSA-CUDA: Multiple sequence alignment on graphics processing units with CUDA. Proceedings of 20th IEEE International Conference on Application-Specific Systems, Architectures and Processors, ASAP 2009. Boston, MA, USA. IEEE. 2009; 121-128.
- Liu Y, Schmidt B, Maskell DL. Parallel reconstruction of neighbor-joining trees for large multiple sequence alignments using CUDA. Proceedings of 23rd IEEE International Sym-posium on Parallel and Distributed Processing, IPDPS 2009. Rome, Italy. Washington: IEEE Computer Society. 2009; 1-8.
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987; 4: 406-425.
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007; 23: 2947-2948.
- Li KB. ClustalW-MPI: ClustalW analysis using distributed and parallel computing. Bioinformatics. 2003; 19: 1585-1586.
- Ebedes J, Datta A. Multiple sequence alignment in parallel on a workstation cluster. Bioinformatics. 2004; 20: 1193-1195.
- Cheetham J, Dehne F, Pitre S, Rau-Chaplin A, Taillon PJ. Parallel Clustal W for PC clusters. Proceedings of the International Conference on Computational Science and Its Applications: Part II, ICCSA 2003. Montreal, Canada. Lecture Notes in Computer Science. Berlin: Springer. 2003; 2668: 300-309.
- Tan G, Feng S, Sun N. Parallel Multiple Sequences Alignment in SMP Cluster. Proceedings of the Eighth International Conference on High-Performance Computing in Asia-Pacific Region, HPCASIA'05; 2005. Beijing, China. Washington: IEEE Computer Society. 2005; 425-431.
- Mikhailov D, Cofer H, Gomperts R. Performance Optimization of Clustal W: Parallel Clustal W, HT Clustal, and MULTICLUSTAL. SGI ChemBio. 2001.
- Aung YL, Maskell DL, Oliver TF, Schmidt B, Bong W. C-based design methodology for FPGA implementation of ClustalW MSA. Proceedings of the 2nd IAPR International Conference on Pattern Recognition in Bioinformatics; 2007. Lecture Notes in Computer Science. Berlin: Springer. 2007; 4774: 11-18.
- Oliver T1, Schmidt B, Nathan D, Clemens R, Maskell D. Using reconfigurable hardware to accelerate multiple sequence alignment with ClustalW. Bioinformatics. 2005; 21: 3431-3432.
- Chaichoompu K, Kittitornkun S, Tongsima S. MT-ClustalW: multithreading multiple sequence alignment. Proceedings of the 20th International Conference on Parallel and Distributed Processing, IPDPS'06; 2006. Rhodes Isl and, Greece. Washington: IEEE Computer Society. 2006; 254-254.
- Liu W, Schmidt B, Voss G, Muller-Wittig W. GPU-ClustalW: using graphics hardware to accelerate multiple sequence alignment. Proceedings of the 13th International Conference on High Performance Computing, HiPC'06; 2006. Bangalore, India. Lecture Notes in Computer Science. Berlin: Springer. 2006; 4297: 363-374.
- Bassoy CS, Torgasin S, Yang M, Zimmermann KH. Accelerating Scalar-Product Based Sequence Alignment using Graphics Processor Units. Signal Processing Systems. 2010; 61: 117-125.
- Hanif MK, Zimmermann KH. Graphics card processing: accelerating profile-profile alignment. Central European Journal of Computer Science. 2012; 2: 367-388.
- NVIDIA Corporation. NVIDIA Corporation, editor. CUDA C Programming Guide Version 4.0. NVIDIA Corporation. 2011.
- Hanif MK. Mapping dynamic programming algorithms on graphics processing units. PhD Thesis, Hamburg University of Technology. 2014.