
1Department of Pharmacology, University of Tennessee Health Science Center, USA
2Department of Psychiatry, University of Cincinnati School of Medicine, USA
3Department of Surgery, University of Cincinnati School of Medicine, USA
4Department of Microbiology and Molecular Cell Sciences, University of Memphis, USA
*Corresponding author: Steven L Parker, Department of Pharmacology, University of Tennessee Health Science Center, 347 Union Avenue, Memphis TN 38163, USA
Received: September 20, 2014; Accepted: October 20, 2014; Published: October 20, 2014
Citation: Parker MS, Wang YY, Sah R, Balasubramaniam A, Sallee FR, et al. Insulin Could Selectively Accelerate Internalization of Slowly Cycling GPCRs. Austin J Pharmacol Ther. 2014; 2 (10).1055. ISSN: 2373-6208.
Phosphorylation of G-protein coupled receptors (GPCRs) is known as the major regulator of sequestration and internalization of these receptors. Several GPCRs have subtypes differing appreciably in internalization rates, which can be importantly related to phosphorylation status of the subtypes. Agents affecting activity of protein kinases and phosphatases could then differentially influence internalization rates for the subtypes. Insulin, as abundant and physiologically pre-eminent protein kinase activator, may stimulate intake of most GPCRs through activation of numerous phosphorylation cascades, with preference for the slowly internalized subtypes. This indeed can be shown with the neuropeptide Y (NPY) Y2 receptor. The intake of this receptor is more than doubled by insulin, while the rate for the fast-internalizing NPY Y1 receptor is increased by insulin less than 20%. Phosphorylation changes triggered by insulin could be of major importance in signaling by less dynamic GPCRs, affecting multiple aspects of their pharmacology and pathophysiology.
Keywords: Protean activity; Protein kinase cascade; NPY Y1 receptor; NPY Y2 receptor
ADAM: A Disintegrin And Metalloproteinase; AT: Angiotensin; EGF: Epidermal Growth Factor; bFGF: Basic Fibroblast Growth Factor; GPCR: G-Protein Coupled Receptor; IGF-I: Insulin-Like Growth Factor 1; IGF-II: Insulin-Like Growth Factor II; NPY: Neuropeptide Y; PYY: Peptide YY; R: Receptor; VEGF: Vascular Endothelial Growth Factor
With G-protein coupling receptors (GPCRs), internalization is a ubiquitous process which achieves the twin results of signal termination and removal of the receptor-attached agonist. A limited constitutive uptake of ligand-free, or uptake of heterologous ligand-propelled receptor may also occur. The uptake of either the receptor or the agonist in some cases could be followed by an intracellular re-use in signaling, or egress by reverse endocytosis (for insulin see [1,2]. However, the internalized receptor appears to be mostly recycled to the plasma membrane, and peptidic agonists typically are degraded.
Several peptidic agonist-driven GPCRs have subtypes that differ considerably in rates of agonist-induced internalization. The slowly internalizing subtypes could owe that status to a variety of causes. As an example, strong adhesion to exocellular partners causes a slow internalization of the neuropeptide Y Y2 receptor [3]. The slowly internalizing subtypes frequently also have lower experimentally proven phosphorylation. This also applies in the case of the Y2 receptor which is not readily phosphorylated, as different from the neuropeptide Y Y1 receptor which has a plethora of easily phosphorylated sites [4]. Internalization of the Y2 receptor is considerably accelerated by insulin, while that of the Y1 receptor is only marginally affected (see Figure 2). The Y1 receptor also has many more high-probability phosphorylation sites as forecast by phosphorylation-predicting programs [5,6].
Among insulin-like factors, pre-eminence of insulin would be based on higher plasma levels enabling a larger volume of interaction with receptors than is attained by insulin growth factor I (IGF-I) and insulin growth factor II (IGF-II). Plasma insulin is essentially free [7], while IGF-I and IGF-II are largely protein-bound [8]. Similar could be expected for most growth factors that work through tyrosine protein kinase receptors. This relates to much weaker ionic character and to interchain disulfides of insulin chains (Table 1 and Figure 1), and the resulting much lower general affinity for proteins. Due to double disulfide bonds between chains A and B, the few ionic residues in insulin are either internally electrostatically engaged, or isolated (Figure 1). The chain A acidic sector via the 31-96 (using the proinsulin numbering) disulfide is facing the basic sector of chain B, leading to internal electrostatic matching. The two disulfides also would reduce access to Glu31 in B and Glu106 in A chain. The C-terminus of chain B contains a counterion “switch” Glu45, Arg46, and Lys53 which is used in insulin oligomerization (see [9]). IGF-I, IGF-II and epidermal growth factor (EGF) have no covalently bound subunits, and their homoionic sectors (which are more prominent than those of insulin) are less internally neutralized, allowing much larger interaction with plasma proteins. Free-form plasma levels of these peptides appear to be below that of insulin by an order of magnitude or more. The use of abundantly available insulin thus should allow for a copious ground layer of phosphorylation events. Insulin binding to either its own receptor or to IGF receptors is much weaker than the binding of IGF-I or IGF-II [10,11]. Additional interactive features (such as lectin complement of IGF-IIR) also may reduce the competitive binding of insulin to IGF receptors. The low affinity of insulin binding would also work to reduce receptor internalization and then also the losses related to cycling.
A note concerning methods for studying insulin effects would underline the need to adequately control the external insulin concentration. In most growth-sustaining media, the rate of external peptide degradation by cell monolayers in wells with stationary medium is considerable. We find that 300 nM PYY(3-36) is ~56% degraded within 24 h by cells in F12/Ham medium with 10% fetal bovine serum (and 10% FBS alone produces about 20% degradation of PYY(3-36) over that period). The experimental medium obviously needs to be refreshed at appropriate intervals (based on kinetics of loss of intact peptide) .
At physiological concentrations (typically reported in the range of 30-300 pM [12,13] ) insulin can increase internalization by driving the phosphorylation and activation of receptor tyrosine protein kinases such as insulin receptor proper [14,15], insulin receptor-like receptor Tyr kinases such as IGF-IR [15-17]}and IGF-IIR [18,19], and even EGFR [20]. Insulin also forms complexes containing insulin receptor, c-src Tyr kinase and Akt Ser/Thr kinase [21]. The above receptors can also be unblocked by ADAM proteinases [22,23], helping transactivation [24]. This would further initiate phosphorylation cascades affecting serine/threonine protein kinases (including especially Akt kinase, but also protein kinase C [25,26]). This would result in phosphorylation of multiple protein classes, including platforms, scaffolds and chaperones involved in various stages of receptor internalization [27-31]. Another protean long-term effect of insulin could be the downregulation of arrestins [32], which however requires prolonged exposure to high levels of insulin.
Recruiting of serine / threonine kinases by insulin should go through phosphorylating services of tyrosine protein kinases directly activated by insulin [21, 25]. Thus, Akt Ser/Thr kinase is activated by PI3 kinase, which in turn is activated by receptor tyrosine protein kinases including EGFR, basic fibroblast growth factor receptor (bFGFR) and vascular endothelial growth factor receptor (VEGFR) [33].
The protean aspects of insulin activity would be enhanced at higher plasma concentrations of insulin. However, phosphorylation of mTORC1 could be accomplished at low nanomolar insulin, with little additional increase at order-of-magnitude higher concentrations [34]. Other targets respond to much higher insulin concentrations [35]. Kinases that act upon intracellular domains of GPCRs could respond to a very large range of activating insulin concentrations, and in most cases would be activated indirectly, by primary insulin responders. The internalization itself also generally depends on receptor dephosphorylation [36,37]. Kinase dephosphorylation could also be important in this regard [38].
Insulin was described as the only hormone required for sustained growth of CHO cells in F12 medium [14]. While the cells may natively express low densities of the insulin receptor, insulin could mainly work through stimulation of non-insulin receptor kinases. This may involve distribution of internalized insulin to various subcellular systems. At less than 10 nM, insulin stimulates kinase activity of mTORC1 complex [34]. Insulin is involved in the basal priming of phosphorylation cascades that drive the endocytosis of most receptors. As an example, low density lipoprotein turnover greatly increases in the presence of only 4 nM insulin [39]. The intake selectivity and specificity of insulin is low. Insulin seems to enter many cell types rather nonselectively. There is evidence for a constitutive, autophosphorylation-independent entry path in CHO cells [40]. However, components of the clathrin system clearly aid internalization of insulin [41].
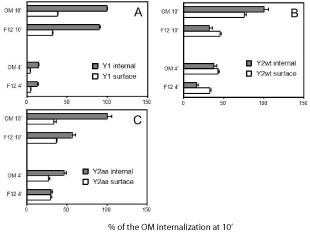
As seen in Fig. 2, Y1 receptor internalization is not importantly driven by insulin in OptiMem medium (graph A). Internalization of the wildtype Y2 receptor was however 2.3-fold larger in OptiMem medium, i.e. at 2 μM insulin (graph B). The stimulation of Y2R intake in this medium was however much larger with the PD34-35AA mutant of the Y2 receptor (graph C), which in CHO cell expressions was shown to internalize more readily than the wildtype receptor [42]. Human holotransferrin at 10 μM increased Y1 and Y2 internalization in either medium by less than 15% (data not shown) . This indicates that internalization of these NPY receptors is not constitutive, unlike e.g. the internalization of the galanin-2 receptor [43].
High insulin in the medium would produce a very quick saturation of any binding sites. The response of the Y2 internalization to insulin may however saturate already at low nanomolar insulin, as seen for internalization of EGF and other growth factor receptors driven by insulin. Responses to high inputs of insulin could even be linked to activation of protein synthesis [44]. However, this would not be important in short-term responses that we have followed. Insulin downregulates EGF receptors at ~2 μM [45], and associates with them at subnanomolar inputs. Clusters of insulin-like growth factor receptors, e.g. IGFR-II, could both inhibit and stimulate transducers, depending on neighboring hydrophobic clusters [46].
Homoionic sectors are the most likely to interact with opposing charge tracts of binding proteins, including receptors [47], and also contain most of basic amino acid residues in all agonists compared in Table 1. As seen in the Table, among short agonists only IGF-II has an ionic cluster, while basic clusters are prominent in VEGF-A. IGF-I and IGF-II have larger homoionic complements than insulin, and should bind counterpart motifs in other proteins (and especially in their specific receptors) at a much higher affinity. This of course is even more applicable to the long growth factors.
To compare degree of phosphorylation and rate of internalization of GPCRs, we used predictions in NetPhos program [5] rather than the phosphorylation values reported in literature, since the experimental conditions producing in situ phosphorylation in many cases differ dramatically, as does the phosphorylation of the same receptor followed by different methods. The upper six pairs of GPCRs in Table 2 are known to strongly differ in the rates of internalization for expressions in the same cell type, the first pair member internalizing faster. Differences for two pairs at the bottom are known to exist, but have not been established in appropriate comparisons. As seen in the Table, five of the six confirmed pairs also differ greatly in the number of highly probable intracellular phosphorylation sites, the faster-internalizing member having many more such sites. The slower member might in some or all cases get accelerated by insulin significantly more than the faster, as we observe for the Y2R compared with the Y1R. However, internalization obviously does not depend on phosphorylation alone. The two angiotensin receptors have very similar predicted (Table 2) and observed [53] phosphorylation. However, the C-terminal tail of angiotensin-2 receptor (AT2R) does not have homoionic clusters and cannot interact with β-arrestin [54], which greatly impairs the intake of this receptor.
Insulin-responding receptors could phosphorylate either the kinases or the chaperones (such as arrestins [55]) that could further act in phosphorylation of the intracellular domains of GPCRs to help internalization of slow-intake receptors, such as the Y2 receptor. However, phosphorylation of the intracellular domains of GPCRs, which in most cases seems to be significantly implemented, is not necessarily critical. An accelerated clearance can be obtained by interaction of phosphorylated platforms /partners and basic clusters in intracellular domains of GPCRs.
Any kinase phosphorylation induced by insulin may have little effect on readily phosphorylated receptors (e.g. The Y1 receptor). Direct phosphorylation of GPCR intracellular domains by the insulin receptor would depend on spatial proximity of the respective domains, and with most receptors with peptidic agonists (which generally have short intracellular loops) could be confined to parts of GPCR “tails” past Helix 8.
The intake of the slowly internalizing receptors could be, as we find for the Y2 receptor, strongly stimulated by high insulin compared to the normally rapidly internalized GPCRs (in the case of the Y2 receptor, the Y1 receptor). Insulin should be acting as a mass action-linked GPCR protein kinase mobilizer. The high concentration of insulin in a medium would provide a very fast kinase mobilization, and likely lead to depletion of the Y2 receptor (or other insulin-sensitive GPCRs) over a prolonged treatment [32,56,57]. This can be expected in hyperinsulinemic states, and may also affect GPCRs that have fast internalization in normoinsulinemic conditions. Any prolonged elevations of plasma insulin may broadly affect levels of many GPCRs, and again especially those with physiologically low internalization rates. This subject should get attention in clinical contexts.
Compared homoionic parameters of some agonists of tyrosine protein kinase receptors.
Peptide |
total residues |
total acidic residues |
number of homoionic acidic |
acidic clusters |
total basic residues |
number of homoionic basic |
basic clusters |
Insulin B |
30 |
2 |
2 |
0 |
4 |
4 |
0 |
Insulin A |
21 |
2 |
2 |
0 |
0 |
0 |
0 |
IGF-1 |
70 |
8 |
6 |
0 |
9 |
8 |
0 |
IGF-2 |
67 |
9 |
8 |
0 |
9 |
6 |
1 |
bFGF |
146 |
15 |
9 |
0 |
28 |
24 |
1 |
VEGF A |
232 |
24 |
13 |
1 |
51 |
50 |
8 |
High-probability phosphorylation sites in intracellular domains and internalization of the subtypes of GPCRs that use long peptidic agonists.
Predictions were made in NetPhos program [5]. Phosphorylation probabilities above 0.9 are considered as highly significant, which for GPCR residues in intracellular domains was experimentally confirmed in a majority of cases.
Receptor |
p > 0.9 intracellular sites |
Internalization reference |
Angiotensin AT1 |
6 |
[48] |
Angiotensin AT2 |
6 |
|
Galanin 1 |
7 |
[43] |
Galanin 2 |
1 |
|
Gonadotropin-releasing 1 |
0 |
[49] |
Gonadotropin-releasing 2 |
7 |
|
Neuropeptide Y Y1 |
6 |
[50] |
Neuropeptide Y Y2 |
2 |
|
Somatostatin 3 |
8 |
[51] |
Somatostatin 4 |
1 |
|
Vasopressin 1a |
9 |
[52] |
Vasopressin 2 |
4 |
|
Neuropeptide FF 1 |
5 |
comparisons needed |
Neuropeptide FF 2 |
1 |
|
Tachykinin 3 |
10 |
comparisons needed |
Tachykinin 2 |
3 |
|
A schematic of insulin molecule. The two internal disulfides link cysteines 31- 96 and 43-109 (numbering as in the proinsulin chain).
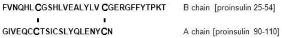
A comparison of internalization of CHO cell-expressed human Y1 and Y2 NPY receptors without and with insulin (2 μM). The labeling was with 200 pM [125I]peptide YY (PYY). The results are relativized to values in OptiMem (OM) medium at 10 min of labeling. Incubation with [125I]PYY was for 4 and 10 min at 37°, starting about 20 min after replacement of the growth medium (F12/Ham, 1:1, at 400 μM geneticin, with 10% fetal bovine serum) by F12 or OptiMem medium with 10% fetal bovine serum. A wildtypeY1 receptor; B wildtype Y2 receptor; C Y2 receptor with P34D35 > A34A35 mutation. The results are averages of 12 samples in three separate experiments.
Austin Publishing Group is an emerging open access publisher specialising in Science, Technology and Medicine is dedicated to serve the biomedical community through its initiatives. Austin Publishing Group is an academic publisher with 100+ peer reviewed open access journals in various subjects such as biomedical, Pharma, Life Sciences, Environmental, Engineering and Management. Austin Publishing Group publishes Open Access eBooks providing free access to vast scientific literature.