
Review Article
Austin Alzheimers J Parkinsons Dis. 2014;1(1): 11.
An Integrative View on Intra- and Inter-Cellular Cooperation Mechanisms in Alzheimer’s Disease
Ancuta Augustina Gheorghisan Galateanu1,2 and Ana-Maria Enciu1,3*
1Department of Cellular and Molecular Medicine, Carol Davila University of Medicine and Pharmacy, Romania
2C. I. Parhon National Institute of Endocrinology, Romania
3V. Babes National Institute of Pathology, Romania
*Corresponding author: Enciu AM, Department of Cellular and Molecular Medicine, Carol Davila University of Medicine and Pharmacy, no8 Eroilor Sanitari Blvd, 050474 Bucharest, Romania.
Received: August 02, 2014; Accepted: September 08, 2014; Published: September 09, 2014
Abstract
Alzheimer’s disease (AD) has been identified as central nervous system pathology more than 100 years ago and for a long time the diagnostic criteria remained the same, based on the anatomopathological findings. Failure of clinical trials in almost every field of AD therapy forced a widening of perspective on molecular pathologies, to the point which, now, AD is considered a multifactorial disease. Recent advancement in AD cell biology uncovered new data regarding amyloid precursor protein and its enzymatic cleavage products, the amyloid beta peptides, such as oligomerization and membrane pore formation. More intracellular deregulated events have been highlighted, e.g. endoplasmic reticulum stress and mitochondrial dysfunction, possibly related through the newly discovered Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM). During the last few years, non-neuronal cell populations came into focus in AD research, such as glial cells, endothelial cells forming the blood-brain barrier or brain non-neuronal stem cells of mesenchymal nature. All these cellular players interact or react to what was considered the central dogma of AD – the amyloid cascade – to the point which this dogma is about to be overthrown.
Keywords: Alzheimer’s disease; Amyloid pores; Endoplasmic reticulum stress; Mitochondrial dysfunction; Blood-brain barrier; Brain mesenchymal stem cells
Introduction
Alzheimer’s disease (AD) has been identified as central nervous system pathology more than 100 years ago and for a long time the diagnostic criteria remained the same, based on the anatomopathological findings: amyloid plaques and fibrillary tangles. For almost half of century no significant progress has been made for this immutable disease, until electron micrograph studies identified amyloid fibrils in both Alzheimer’s disease and Down syndrome patients [1]. Combined with genetic studies, the Amyloid Precursor Protein (APP) gene and splicing proteins were identified and soon, familial forms of the disease and subsequent mutations were reported [2-4]. Familial studies, as well as molecular studies of late-onset AD led to identification of other risk factors, such as apolipo protein E [5,6]. The new wave of knowledge came from attempts to identify the proteases involved in fibril generation and AD pathology. Observation that mutated presenilins (enzymes involved in Notch signaling) are related to aggressive forms of early onset AD [7] led to a demonstrated relationship between these gamma secretase and APP [8] followed by identification of alpha secretase [9] and beta secretase [10]. Beta secretase has soon become the main point of interest in the development of an efficient, targeted, anti-AD therapy [11], but failed to fulfill the expectations during the next twenty years. Failure of clinical trials in almost every field of AD therapy forced a widening of perspective on molecular pathologies, to the point which now, AD is considered a multifactorial disease. Genome-wide association studies have identified during the last 5 years involvement of cholesterol metabolism, endosomal system and innate immunity in AD, albeit at frequencies below 50% and modest impact on risk [12]. Although most in vitro studies used neuronal cell populations, lately, new cell types (microglia, endothelial cells) were shown to contribute to, or be influenced by molecular pathogenesis of AD. Parallel advancement in other types of dementias, such as vascular dementia, raised the possibility that pathologic and diagnostic limits may not be so clearcut. Far from been exhaustive, the present review addresses recent advances in several fields of neuroresearch related one way or the other to AD, from intracellular, compartment-specific processing of AD related proteins, to types of cells of importance in the progression of the disease.
Intracellular Pathogenic Links
Amyloid cascade and tau hyperphosphorylation- a 5 years update
Amyloid cascade: Amyloid cascade is a two-step enzymatic cleavage, initiated normally by a class of proteases (ADAM), also called a secretases, that shed the extracellular domain of APP and generate a C-terminal fragment, further cleaved by a gamma-secretase complex, with presenilins as main enzymatic component. In AD, a β secretase cleaves the extracellular domain of APP, generating the C99 fragment and creating the premises to yield Aβ peptides 40 and 42, with the latter exhibiting high propensity to aggregate in the extracellular space and create amyloid plaques. Besides these two main Aβ species, several other truncated species have been identified Cerebrospinal Fluid (CSF), plasma or interstitial fluid [13]. Very recently, through nuclear magnetic resonance and electron paramagnetic resonance spectroscopy, it was shown that C99 has a flexible transmembrane domain with a binding site for cholesterol [14]. It is now known that amyloid peptides are detrimental to synaptic activity even inoligomeric forms [15], or intracellular deposits [16]. Up to this moment, all three classes of secretase are fairly well described along with the secretory pathway, alternative enzymatic cleaving sites and subsequent peptide products. Cleavage occurs with optimum efficiency in the endosomal compartment, characterized by a lower pH than the rest of cellular compartments.
Amyloid precursor protein and cholesterol metabolism: Although the relationship between amyloid plaque and cholesterol has been long reported, only recently, functional cholesterol-binding domains in several amyloidogenic proteins have been identified. For APP, although cholesterol binding domains are found in both C99 and Aβ peptides, they are only partially overlapping, leading to greater affinity of cholesterol for Aβ. Furthermore, cholesterol binding enables Aβ peptides to form membrane pores by oligomerization. This data offered the perspective of an original therapeutic strategy using cholesterol competing-drugs, such as bexarotene for the treatment of Alzheimer’s and other neurodegenerative diseases that involve cholesterol-dependent toxic oligomers [17].
But not only cholesterol and apolipo proteins are shown to influence Aβ deposition and cognition, recently other players involved in cholesterol metabolisms were studied and reported to exert varia influences on AD pathology.
Hemizygosity of ATP-binding cassette transporter A1 (ABCA1) transporter (that regulates cholesterol efflux and formation of High- Density Lipoprotein (HDL)) increases Aβ deposition, but only when associated with ApoE4 phenotype [18].
In turn, overexpression of human apoA-I in the circulation prevents learning and memory deficits in APP/PS1 mice, but apparently without changing the burden of Aβ deposition in the brain. The protective effect was supposedly due partly to anti neuroinflammatory effect and diminished cerebral amyloid angiopathy [19].
Hormonal signaling: AD has become today the “diabetes of the brain”, or type 3 diabetes, due to its well characterized resistance to insulin signaling, accompanied by IGF-1 resistance and closely associated with IRS-1 dysfunction, potentially triggered by Aβ oligomers [20].“Resistance to insulin” in AD brain has grossomodo the same significance as in diabetes – defective downstream insulin signaling, in spite of sufficient hormone levels. Mechanisms responsible for brain insulin resistance, reported so far are: i) increased IGF-1R levels with aberrant distribution in AD cortex [21]; ii) phosphorylation of insulin receptor substrate 1 and 2 (IRS 1/2) [22], acting a negative feedback exerted by various kinases (ERK2, glycogensynthase kinase–3 (GSK-3), mammalian target of rapamycin/ S6K1 (mTOR/S6K1)and certain isoforms of Protein Kinase C (PKC)) and from feed-forward inhibition exerted by I?β kinase β (IKKβ) and JNK1/2 [20]; iii)reduced cytosolic and/or membranous levels of PI3K [23]; iv) reduced GLUT-1 expression at blood-brain barrier in AD patients [24].
In turn, early hyperinsulinemia is enough to exacerbate AD pathology observed in APP/PS1 mice [25]. Intranasal administration of insulin seems to improve cognition in both healthy and MCI patients [26].
Circulating leptin was associated with a reduced incidence of dementia and AD [27], but excess Aβ can potentially lead to a pathologically low leptin state, early in the disease process, that progressively worsens as the amyloid burden increases [28].
Anti-pathogen activity of Aβ: Possible roles played by pathogens in AD were repeatedly reported, from viral [29,30] to bacterial [31] and fungal [32] infections, as well as in other neurodegenerative diseases, such as Parkinson’s Disease and amyotrophic lateral sclerosis [33]. Most notably, recent evidence indicate the herpes simplex virus-1 to be a strong presence in the pathogenesis of AD, leading to generation of multiple APP fragments with neurotoxic potentials [34], induces accumulation of intracellular Ca(2+) and downstream signaling to Ca(2+)-dependent APP phosphorylation and intracellular accumulation of Aβ42 [35] and abnormally hyperphosphorylated tau [36].
In 2010, Soscia et al proposed Aβ as an antimicrobial peptide and proved it to have antimicrobial effect in vitro, comparable with LL-37, an archetypical human antimicrobial peptide. The authors also showed that brain homogenates of AD patients have higher antimicrobial activity than aged matched non-AD samples and that antimicrobial action correlates with tissue Abeta levels [37]. Further on, other peptides generated by enzymatic c leavage from the extracellular domain of APP exert antimicrobial effects in vitro against Gram-negative and Gram-positive bacteria, putatively via a membrane permeabilising activity [38]. Aβ42 was also shown to reduce uptake of IAV by epithelial cells and appeared to possess direct antiviral effect, possibly by promoting viral aggregation and further helping of phagocytic viral clearance [39].
Tau hyperphosphorylation: The second main pathogenic link involved in AD is hyperphosphorylation of tau – a microtubule associated protein, involved in cytoskeleton stability. Unlike APP, able to induce by itself a complete AD phenotype (both amyloid plaques and tau hyperphosphorylation) when overexpressed in animal models, abnormal tau phosphorylation leads to tautopathies. In AD tau pathology is most likely secondary to amyloid cascade, the main link between the two being glycogen synthase kinase 3, activated by the Aβ peptides via insulin and Wnt signaling pathways [40].
Intraneuronal interaction between Aβ peptides and tau has been proposed more than 5 years ago to occur on multiple tau peptides, especially those in exons 7 and 9, enhancing tau phosphorylation by GSK3beta [41] and reconfirmed lately in postmortem brains from AD patients at different stages of disease progression and animal models [42]. Furthermore, recent evidence suggests that tau protein may mediate amyloid-β peptide (Aβ) toxicity by modulating the tyrosine kinase Fyn [43]. Animal models with a heterozygous Fyn phenotype showed increased soluble Aβ accumulation and worsened spatial learning in the absence of changes in tau phosphorylation [44]. Tau-Fyn interaction has been recently investigated in order to map Fyn binding site on Tau and target interaction inhibitors by highthroughput screening [45]. Phosphorylation of tau is also influenced by Cdk5, a cyclin kinase which, unlike its other family members is not involved in cell cycle progression but in phosphorylation of cytoskeletal proteins and synatpic formation. In AD was reported as one of the major players causing aberrant hyperphosphorylation of tau through phosphorylation of specific repeats in neurofilaments of heavy and medium molecular weight [46]. Silencing of CDK5 reduces the phosphorylation of tau in primary neuronal cultures and in the brain of wild-type mice [47]. Recent data report, however, that there is a possibility that a therapeutic effect would be attainted only by dual kinase inhibition [48].
Another recent input on tau biology is the relationship between the Unfolded Protein Response (UPR) and early tau phosphorylation [49] as well as tau involvement in mitochondrial dysfunction [50]. Truncated tau induced significant mitochondrial fragmentation in neurons and when combined with Aβ at sublethal concentrations, increased the levels of oxidative stress. It also enhanced Aβ-induced mitochondrial potential loss in primary neurons [51].
Tau phosphorylation seems to be modulated also by neurotransmitters input, as specific serotonergic denervation increased tau phosphorylation in denervated cortex, without affecting amyloid-beta (Aβ) pathology [52].
Posttranslational control of main players in AD by microRNAs
MicroRNAs are small non-coding RNAs, than can pair, perfectly or imperfectly, with the 3’ untranslated region (UTR) of different mRNAs. A perfect complementarity between the mRNA and miRNA will lead to mRNA degradation, while an imperfect match will stop the protein translation. Either way, microRNAs act as downregulators of protein synthesis, for one, several or, in some cases, tens of mRNAs. It is also possible for one mRNA to be targeted by several different microRNAs that regulate its expression at different time points in the life of the cell, or under different environmental conditions.
As this posttranslational regulatory pathway has been identified no more than two decades ago, the bulk of data regarding the main actors in AD pathology (APP, beta and gamma- secretases) regulated by microRNA is no older than 5-10 years. The research in the field of microRNA involvement in AD pathology is expanding, along with discovery of new microRNA sequences. Up-to-date, in one microRNA data base (https://www.mirbase.org) are listed over 1800 sequences of identified microRNAs in humans, out of which many species or clusters are expressed in the central nervous system in a both timedependent manner [53] and cell-specific manner [54]. Many of micro RNAs expressed in the human brain are not conserved between different families of primates, suggesting that, phylogenetically, they are a recent acquisition [55].
From the full range of miAR Nuri identified in the brain, so far only a few have been associated with neuro degenerative processes: miR-133b [56], miR433 [57] in Parkinson’s disease, miR-9 in Huntington’s disease [58,59], miR-132, miR-124a, miR-125b, miR- 107, miR-219 andmiR-128 in Alzheimer’s disease [60-62].
One of the first reports of a miRNA profile modification in patients with AD belongs to Walter Lukiw, studying human hippocampus from fetal and adult patients with AD. The results indicate the abnormal expression ofmiR-9, miR-125b andmiR-128 in the hippocampus of AD brains [61]. Another study by Wang W X et al. On AD patients’ brains, compared with control subjects without cognitive impairment, showed a marked decrease of miARN-107, even in the early stages. Wang et al. demonstrated that the 3’-UTR sequence of BACE1 mRNA is a possible site of attachment of them iARN-107, regulating the expression of the enzyme. In addition to the decrease observed in the cortex of the temporal, the same trend was observed in the motor cortex. These changes indicate a global change miR-107 levels in the whole brain, even in area sun affected by AD pathology [63] . The 3 ‘UTR of mRNA of BACE1 is also targeted by miRNA-29a/b-1cluster, as decreased expression of these microRNAs was demonstrated in patients withsporadicAD. Consistent with the literature [60,64-68] an increase of BACE1 expression was demonstrated through quantitative RT-PCR. Decreased expression miR-29a/b-1 and increased BACE1 is not specific to a particular cortical area, as demonstrated by cerebellum assessment [69]. Cogswell metal, Quantified the expression of over 300 miRNA sites in the hippocampus, medial front algyrus and cerebellum taken from different stages of AD compared with agematched controls. These data show modifications of certain species of miRNA – out of which miR-9 and miR-132 were repressed in the hippocampus and frontal gyrus, correlating with the progress and location of pathological lesions. In addition they report detecting miRNAs in the CSF, with different levels from controls [70]. Over expression of miR-125b causes tau hyperphosphorylation and targets the phosphatases DUSP6 and PPP1CA and the anti-apoptotic factor Bcl-W [71], whereas miR-922 increased the levels of phosphorylated tau by regulating ubiquitin carboxy-terminal hydrolase L1 [72].
Recently, other species of miRNAs are found dysregulated in animal models of AD, related not to amyloid cascade but to other pathogenic links such as synaptic plasticity (upregulation of miR-181 [73]), or immune-related (miR-155, upregulated simultaneously with an increase of microglia and astrocyte activation [74]).
Mitochondrial dysfunction
Alteration of mitochondrial metabolism in AD patients has been well documented in the literature and confirmed in in vitro studies showing that Aβ affects mitochondrial DNA and proteins, leading to impairments of the Electronic Transport Chain (ETC) and ultimately mitochondrial dysfunction [75]. In a triple transgenic mouse AD model, mitochondrial dysfunction was detectable from embryonic stage, continues throughout the reproductive period and is exacerbated during reproductive senescence, unlike control wild type littermates, in which oxidative stress and a significant decline in mitochondrial function was demonstrated only with reproductive senescence [76]. A significant decrease in mitochondrial membrane potential was also noted in two cell models of AD, also accompanied by a decrease in ATP synthesis [77]. Interesting results have also been reported in selected nerve cell lines [78], along with already consecrated in vitro neuronal models PC-12 [79] and SHSY-5Y [80] cell lines
The relationship between mitochondrial dysfunctions and other pathogenic links in AD was under intense scrutiny, most efforts concentrated on relationship between this organelle and aberrant APP processing. Correlating with the newly demonstrated ability of Aβ to oligomerize at cell membrane site, increasing evidence suggested that both APP and Aβ peptides accumulate also in mitochondrial membranes, forming pores that increase permeability and further promote the excess accumulation of Ca(2+) [81]. Both full length APP and C99 were shown to target to the mitoplast (inner membrane and matrix compartments) in brains of an AD transgenic mice, which seemed to be almost completely dependent on BACE 1 activity [82]. As for Aβ peptides, they were found at mitoplast level in both monomeric and oligomeric forms, even as early as 2 months of age in transgenic AD mice, generating free radicals, ultimately leading to oxidative damage [75]. Mass spectrometry studies identified heat shock protein 60 (HSP60) to be responsible for Swedish mutated APP (KM670/671NL) translocation to mitochondrial matrix, along with critical components of gamma-secretase complex [83].
Studies on the integrity of the inner mitochondrial membranes and functional status of electron transfer complexes reported that APP treatment negatively influences the complex IV activity, while the activities of complexes I and II did not change. Furthermore, activity of complex III was significantly enhanced in APP expressing cells, as compensatory response, in order to balance the defect of complex IV, compensatory mechanisms that was, however, unable to prevent the strong impairment of total mitochondrial electron transport chain [84].
Another pathogenic interaction proposed recently to explain the mitochondrial dysfunction under AD burden was upregulation of voltage-dependent anion channel 1 protein (VDAC1) levels, found in the cortical tissues from the brains of patients with AD, relative to controls, as well as in transgenic laboratory mice [85].
The association between mutant APP and mitochondrial dysfunction in not cell-type specific, as it has been very recently reported to also occur in the striated muscle fibres of a transgenic animal used as an AD model [86].
Last, but not least, mitochondrial dynamics have also been reported to be altered in AD, with a shift of balance towards organelle fission and decreased mitochondrial anterograde movement, to accompany the defective functions [87]. Furthermore, downregulation of Akt signaling, also possibly related to insulin resistance in AD, was shown to diminish mitochondrial biogenesis, with subsequent memory impairment in laboratory animals [88]. Another pathologic link relating mitochondria and AD is Aβ-binding alcohol dehydrogenase (ABAD) - a direct molecular link from Aβ to mitochondrial toxicity. Aβ was shown to interact with ABAD in the mitochondria of AD patients and transgenic mice overexpressing ABAD, with the latter manifesting exaggerated neuronal oxidative stress and impaired memory in an Aβ-rich environment [89].
Endoplasmic reticulum stress in AD
Endoplasmic Reticulum (ER) stress is manifested by several functional dysregulations such as calcium release or accumulation of unfolded proteins in the lumen of the organelle. This loss of homeostasis is sensed by specific transmembrane proteins that trigger the Unfolded Protein Response (UPR) – three different signaling pathway that regulate gene transcription to increase chaperone levels into assisting ER to properly fold proteins, or even to downregulate production of mRNA of unfolded proteins [90] (Figure 1).
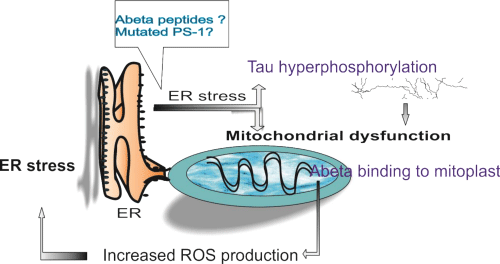
Figure 1: Endoplasmic reticulum – mitochondrion cooperation in Alzheimer’s disease. Abbreviations: ER: Endoplasmic Reticulum; PS: Presenilin; ROS: Reactive Oxygen Species.
At first, endoplasmic reticulum was related to AD pathogenesis through mutated presenilins and perturbed calcium release [91]. Soon enough was shown that mutated PS-1 downregulates the Unfolded Protein Response (UPR) signaling in ER, possibly through inhibited activation of stress transducers at the ER membrane [92]. In sporadic ADexperimental data showed that the aberrant splicing isoform of PS-2 gene, generated by exon 5 skipping, is responsible for downregulation of the signaling pathway of the UPR, in a similar fashion to that reported for mutants of PS1 linked to familial AD. Prolonged ER stress leads ultimately to apoptosis, which in AD neurons was mediated by caspase-4 [93].
The relationship between ER stress and pathologic accumulation of Aβ peptides and hyperphosphorylated tau protein in the AD brain is still under study, as controversial results have been so far reported by different group studies (reviewed in [94]). ER stress triggered by Aβ was shown to promote cholesterol synthesis and mitochondrial cholesterol influx, resulting in mitochondrial glutathione depletion [95]. Sustained activation of the endoplasmic Reticulum (ER) stress response by amyloid-beta (Aβ) peptide was shown to induce apoptotic cell death not only in neurons but also in brain endothelial cells [96]. Downregulation of ER membrane transducers was confirmed in AD human brains and an interesting hypotesis was recently proposed, that linked one of this transducers with the expression of an α secretase (ADAM10) involved in non-amyloidogenic processing of APP [97].
Not only APP seems to be linked to ER stress, but also tau protein and its level of hyperphosphorylation. A strong association between the presence of UPR activation markers and p-tau was observed in the hippocampus of front temporal dementia cases, occurring independently from Aβ deposits [98]. In turn, it was proposed that tau increased the levels of ubiquitinated proteins in the brain and triggered activation of UPR by interfering with protein quality control in the ER. Increased levels of ubiquitinated proteins were accompanied by increased levels of UPR activation [99].
Non-Neuronal Cells Involved in AD Pathogenesis
Microglial activation and oxidative stress
Based on current data one cannot establish whether inflammation is a cause, a promoter, or a secondary phenomenon in AD [100], but with accumulating evidence for oxidative stress as an important pathogenic factor in AD, theories have emerged speculating that it is involved in the initiation of the disease. In fact, oxidative damage was found to be one of the first events in the disease progression [101], it has been highlighted in MCI and AD patients and it gets more intense as the disease advances [102]. An increase in microglial activation has been observed in very early stages of AD, with a number of studies reporting aggregation of activated microglia around amyloid plaques in animal [103] and human brain [104-106] and, interestingly, it does not seem to be preserved over time [107]. Inflammation-related gene profiling in AD indicate microglia to be in an alternative, “reconstructive activation state, rather than the classical, destructive state” [108], but in the complex environment of neuroinflammation, there is not a clear cut between “destructive” and “regenerative” actions of microglial subpopulations. The neuroprotective effects of activated microglia in inflammatory environments are partially overlapping its phagocytic, destructive ones and may be triggered by signalling molecules produced by apoptotic neurons [109].
Like most of AD pathogenic links, there seems to be a dual relationship between reactive oxygen species and Aβ accumulation. Aβ-stimulated microglia produce and secret proinflammatory molecules and neurotoxic factors [110], such as IL-1a, IL-8 and TNFa, that were proposed as biomarkers able to distinguish AD from controls [111,112]. In addition, certain peptide fragments (such as, for example, Aβ 25-35) have the intrinsic capacity of peroxidizing membrane lipids and generate typical products such as 4-hidroxinonenal (HNE) [113] – aldehyde with proven ability to interfere with membrane ATPase and ion channels, including those involved in calcium homeostasis. Furthermore, HNE is involved in altering the metabolism of glucose via neuronal GLUT-3 transporter and the GLUT-1 astrocytic isoform. Impaired glucose metabolism is proven by studies FDG-PET (18F-2-fluoro-deoxy-D-glucose) and is a common denominator for all changes in cognition from MCI and vascular dementia up to AD. An interesting correlation can be made with the results of PET studies in MCI and patients with possible or probable AD, which showed a decrease in glucose transport into nerve tissue, with hippocampal deficit level as a predictor of evolution to AD [114]. In turn, hydroxyl radicals can react with Aβ, triggering enhanced oligomerization and aggregation [115].
Astrocytes
Astrocytes are the most numerous cells in the CNS, involved in maintenance of neuronal homeostasis. They have been proposed to be part of the proper synaptic signaling ( in the so called tripartite synapse [116]). In vitro, human astrocytes are highly sensitive to oxidative stress and trigger a senescence program when faced with multiple types of stress. One study showed that the frontal cortex of AD patients harbored a significantly greater burden of senescent astrocytes compared with non-AD age-matched controls. These astrocytes grown in vitro released a number of inflammatory cytokines and were, in turn, stimulated into senescence by Aβ (1-42) [117]. Aβ treatment is directly able to induce intracellular calcium transients and spontaneous intercellular calcium waves in isolated astrocytes in purified cultures, even at low concentrations [118], inducing alterations in astrocyte cooperation, which is known to occur through connexins-organizing gap junctions. In the brain of AD transgenic mice, an overexpression of connexins 30 and 43 were observed around amyloid plaques [119], supporting the abnormal activation of astrocytic signaling in AD. Conversely, astrocytes appear to be involved in the clearance of Aβ (1-42) [120].
Astrocytes, along with oligodendrocytes are also a source of APP [121] and inflammatory cytokines increase levels of endogenous BACE1, APP, and Aβ and stimulate amyloidogenic APP processing in astrocytes and neurons (Figure 2). The same effect was observed under oligomeric and fibrillar Aβ 42 treatment [122], creating a viciouscircle in the AD brain.
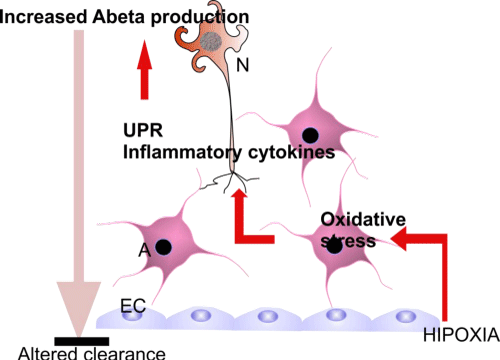
Figure 2: Cooperation and pathogenic links between cell types in AD brain. Abbreviations: EC: Endothelial Cells; A: Astrocytes; N: Neurons.
Endothelial cells and blood-brain barrier
BBB impairment is the common denominator of many neurological disorders, whether purely neurological, or systemic. Alzheimer’s disease is not an exception and BBB alteration hypothes is emerged from observing Congo Red positive amyloid deposits in cerebral microvasculature [123].
One hypothes is suggests that increased production of Alzheimer’s disease peptide exceeds the capabilities of clearance of nervous tissue, including transportation through the BBB. The CSF of healthy individuals contains picograms of Aβ peptides, which are increased in AD patients, especially the 42 amino acid form, which seems to confirm the existence of an alteration of the BBB or at least a lack of transport through the BBB. Studies in patients with AD showed an increase in Aβ peptides bound to lipoproteins and serum proteins, what might indicate a correlation between brain deposits and the circulating peptides distribution. The importance of the blood stream as a source of Aβ peptides is highlighted by studies of transgenic mice in which the treatment with a peptide chelating agent would reduce the plasma and cerebral amyloid load, by virtue of the equilibrium maintained between the two compartments by specific transport mechanisms [124].
Altered BBB permeability holds, from this perspective, a major role in disrupting the blood-brain circuit of the neurotoxic peptide Aβ42. Amyloid precursor protein exists as a membrane receptor in circulating blood cells and blood platelets membrane as well as in soluble circulating form. Opening BBB initiates pathological parenchymal amyloid deposition and neuro toxic mechanisms that would result in the destruction of neurons sensitive to ischemia, mostly hippocampus granular neurons. Is chemicinjury, leading to BBB breakage, is associated with increased expression of β-secretase, which tilts the balance in favor of the amyloid ogenic pathway. This result is associated with the demonstrated accumulation the Ct of the APP fragments in cell-free amyloid plaques, in animal models of focal cerebral in farction [125].
Aβ is cleared through the BBB using a pair of transporters: RAGE (receptor for advanced glycation end products) and the soluble LRP1 (LDL Receptor-related Protein1), which under normal conditions operates as a chaperon protein for the Aβ peptide in plasma and is responsible for its transport to the liver where it is metabolized. In patients with AD, LRP1 is found in oxidized form, which altersitsbinding ability. Transport rate is influenced by ApoE, that correlates with increased risk of AD in carriers of isoform e4. It has already been shown that ApoE plays a role as Aβ peptide chaperone and carrier so fallele 2 and 3 have a rate of transport through the BBB significantly higher than the allele 4 carriers [124,126]. The transport can also be increased by neuroinflammatory cytokines TNF and IL-6, which contribute to the inflammatory micro-environment and participate at the same time to increased BBB permeability. Another possible modulator of transport by RAGE/LRP1 is the endothelial NO synthase, whose level is reported to below in the brains of AD patients [127].
Another clearance mechanism at the BBB involves glycoprotein P (Pgp) [128], which seems to be decreased in amyloid angiopathy at endo the lial level. Although considered in conclusive, there were reports that the inverse relationship between Pgp expression and amyloid deposition is related particularly to Aβ40 [129].
Kinetic analysis of Aβ distribution across the mouse BBB showed that most of Aβ40 is cleared across BBB. Clearance rates differ, however, between mouse and human models, but the ratio between transport and local degradation is maintained in favour of BBB clearance [130].
AD endothelial dysfunction has been reported in vascular defects of organization at the microscopic level-increased amount of collagen IV, laminin and proteoglycans in the vascular basal lamina, reduced vascular bed [131]. Vascular changes in the AD brain are also reflected in aberrant angiogenesis. Vessels of neoformation, fenestrated, with more permeable tight junctions, morphological abnormalities and changes in endo the lial basal lamina were described in the brain of AD patients. The diameter of vascular branch in gand abnormal basal lamina abnormalities have been reported in animal models of AD brain by Nakajima [132] and Meyer [133]. Also demonstrated in the brains of AD patients, over production of angiogenic mediators (Angiopoietin-2 and VEGF) may contribute to vessel formation. VEGF is produced by astrocytes in response to hypoxia, which, as mentioned above, it has been proposed to link pathogenic and there have been studies reported high levels of VEGF and TGFβ in CSF of patients diagnosed with probable AD. Note that the mere presence of VEGF does not necessarily indicate angiogenesis, as this cytokine is involved in axonal growth, cell survival, chemotaxis form macrophages and granulocytes. However avβ3 integrin positivity in patients with AD does, as documented by Desaietal in the hippocampus of demented patients compared with healthy subjects [134].
Recent data indicate that Aβ treatment of monolayered cells (such as epithelial or endothelial cells) disrupts monolayer integrity in term of tight junction proteins [135] or Aβ transport across the cell [136].
Neural stem cells and stem cell niche in AD
Regarding neurogenesis in AD brain, the perspective is dual, but it is usually approached, in original articles, form a unilateral point of view: 1- is there any adult neurogenesis activity left in the AD brain? And 2- how does the already altered AD-related microenvironment influences the neurogenesis of an already aged brain.
By now, the status quo of neurogenesis in aged brain states that neurogenesis is preserved, albeit at lower rates than in the healthy adult and even more so in the AD brain [137]. Although reports considerably differ depending on the model used and investigated marker [138], the trend seem to indicate that in AD, the neurogenesis reserve is activated, meaning there is a higher rate of division of neural stem cells and a higher proportion of committed progenitors in subventricular zones and hippocampal dentate gyrus, but these progenitors fail to complete their differentiation program [139] and engage into the rostral migratory stream
However, in vitro studies using human neurospheres reported, unlike in vitro models using rodent NPCs, that Aβ 1-40 treatment impaired proliferation and differentiation of precursor cells [140]. Results of Aβ effects reported on mouse brain-derived neurospheres differ, however, with the type of peptide used: i) Aβ 25-35 induces neuronal differentiation and apoptosis in neural committed cells [141]; ii) Aβ40 promotes neurogenesis in NPCs [142]; iii) Aβ42 stimulates neurosphere formation and increases the number of neuronal precursors [143]; it also has a reported effect of inducing astrocytic differentiation [142].
Also, advanced stages of the disease impaires proliferation and survival of Neural Precursor Cells (NPC) in mouse brain [144] and the decrement in NPC number was correlated with accumulation of Aβ, even in oligomeric, diffusible form [145]. Although Kolecki et al. confirmed the previous results, they reported that overexpressing APP and Aβ in transgenic mice do not interfere with the mitotic activity of NPC, as assessed by Ki-67 [146]. Other reports showed that fibrillar Aβ-42 seems to be involved in hindering neurogenesis either by generating an inappropriate environment for neuroblasts to mature, or inducing senescence of neuronal stem or progenitor cells [147]. Exposure of NSCs to amyloid-β oligomers lowers their dividing potential, favours gliogenesis and attenuates mobility [148].
Neural stem cells and progenitor cells can be cultured from rapid autopsy samples of SVZ from elderly human subjects, including patients with age-related neurologic disorders and maintained in vitro to generate neurospheres [149]. Interestingly, the percentages of β-tubulin (III)+ cells differentiated from AD and control SVZ neurospheres using a neuron-promoting differentiation protocol were equivalent, promoting the idea that committing abilities of NSC are not modified by the course of the disease.
Brain mesenchymal stem cells
Although initially described as a distinct subpopulation in the haematopoietic bone marrow stroma, Mesenchymal Stem Cells (MSC) have been also identified in dermis, adipose tissue, skeletal muscle and tendons [150] and recently, in the brain, in relationship with brain vasculature. Brain mesenchymal stem cells express mesenchymal (CD105, CD13) and pericyte markers (PDGFR-β) along with, a-SMA, NG2, CD13, CD49d, CD73, CD90, CD106 andRGS5 [151]. They are multipotent, showing adipocytic, chondrocytic and ostocytic activity, under appropriated stimuli, but also, when exposed to glial induction medium, were able to differentiate into oligodendrocytes or astrocytes. They are also able to generate an immature neuronal phenotype [151].
One MSC particular feature is autophagy regulatory digestion pathway for aggregated proteins and organelles that cause various neurodegenerative diseases, such as a synuclein in PD or tau deposits in tauopathies and AD. Unlike NSCs, MSCs possess immunomodulatory functions, are able to influence activation of microglia to an alternative phenotype, as part of the repair process and extracellular matrix reorganization, not accompanied by production of pro-inflammatory cytokines [152], increase microglialrelated Aβ clearance, reduce tau hyperphosphorylation and improve spatial learning and memory impairments in MSC transplanted AD animal models [153].
Future perspectives
The intricacies of AD pathological links seem to be increasing, as the perspectives widens in attempt to a better understanding of the disease. The deeper one looks inside the cell, more correlations will be brought to surface, out of which the significant, disease-modifying ones have to be highlighted. Therapeutic pipelines still address classical features of AD (BACE and ? secretase inhibition, prevention of aggregation of hyperphosphorylated tau) and we expect to see, after a decade of failures and accumulated knowledge, some viable results in the near future. In the meanwhile, new pathogenic links are being addressed; some of them benefiting from already tested and approved drugs, such as insulin therapy.
Nevertheless, the bulk of data contributes also to advancement in normal cell biology of APP, which will lead, in turn, to a better grasp of pathologic alterations.
Acknowledgment
This work was partially supported by the Sectorial Operational Programme Human Resources Development (SOPHRD), financed by the European Social Fund and the Romanian Government under the contract number POSDRU 141531.
References
- St George-Hyslop PH, Tanzi RE, Polinsky RJ, Haines JL, Nee L, Watkins PC, et al. The genetic defect causing familial Alzheimer's disease maps on chromosome 21. Science. 1987; 235: 885-890.
- Mann DM, Jones D, Snowden JS, Neary D, Hardy J. Pathological changes in the brain of a patient with familial Alzheimer's disease having a missense mutation at codon 717 in the amyloid precursor protein gene. Neurosci Lett. 1992; 137: 225-228.
- Mullan M, Houlden H, Windelspecht M, Fidani L, Lombardi C, Diaz P, et al. A locus for familial early-onset Alzheimer's disease on the long arm of chromosome 14, proximal to the alpha 1-antichymotrypsin gene. Nat Genet. 1992; 2: 340-342.
- Lannfelt L, Bogdanovic N, Appelgren H, Axelman K, Lilius L, Hansson G, et al. Amyloid precursor protein mutation causes Alzheimer's disease in a Swedish family. Neurosci Lett. 1994; 168: 254-256.
- Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimer's disease. Lancet. 1993; 342: 697-699.
- Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993; 43: 1467-1472.
- Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997; 3: 67-72.
- Xia W, Zhang J, Ostaszewski BL, Kimberly WT, Seubert P, Koo EH, et al. Presenilin 1 regulates the processing of beta-amyloid precursor protein C-terminal fragments and the generation of amyloid beta-protein in endoplasmic reticulum and Golgi. Biochemistry. 1998; 37: 16465-16471.
- Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, et al. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998; 273: 27765-27767.
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, et al. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999; 402: 533-537.
- Vassar R. The beta-secretase, BACE: a prime drug target for Alzheimer's disease. J Mol Neurosci. 2001; 17: 157-170.
- Franco R, Cedazo-Minguez A. Successful therapies for Alzheimer's disease: why so many in animal models and none in humans? Front Pharmacol. 2014; 5: 146.
- Kummer MP, Heneka MT. Truncated and modified amyloid-beta species. Alzheimers Res Ther. 2014; 6: 28.
- Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, et al. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science. 2012; 336: 1168-1171.
- Kayed R, Lasagna-Reeves CA. Molecular mechanisms of amyloid oligomers toxicity. J Alzheimers Dis. 2013; 33 Suppl 1: S67-78.
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. 2007; 8: 499-509.
- Di Scala C, Chahinian H, Yahi N, Garmy N, Fantini J. Interaction of Alzheimer's β-amyloid peptides with cholesterol: mechanistic insights into amyloid pore formation. Biochemistry. 2014; 53: 4489-4502.
- Fitz NF, Cronican AA, Saleem M, Fauq AH, Chapman R, Lefterov I, et al. Abca1 deficiency affects Alzheimer's disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J Neurosci. 2012; 32: 13125-13136.
- Lewis TL, Cao D, Lu H, Mans RA, Su YR, Jungbauer L, et al. Overexpression of human apolipo protein A-I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease. J Biol Chem. 2010; 285: 36958-36968.
- Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012; 122: 1316-1338.
- Moloney AM, Griffin RJ, Timmons S, O'Connor R, Ravid R, O'Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer's disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging. 2010; 31: 224-243.
- Talbot K, Han L, Schneider JA, et al. O3-02-02. Alzheimer's & Dementia. 2006; 2: S54.
- Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012; 122: 1316-1338.
- Mooradian AD, Chung HC, Shah GN. GLUT-1 expression in the cerebra of patients with Alzheimer's disease. Neurobiol Aging. 1997; 18: 469-474.
- Ramos-Rodriguez JJ, Ortiz-Barajas O, Gamero-Carrasco C, de la Rosa PR, Infante-Garcia C, Zopeque-Garcia N, et al. Prediabetes-induced vascular alterations exacerbate central pathology in APPswe/PS1dE9 mice. Psychoneuroendocrinology. 2014; 48: 123-135.
- Freiherr J, Hallschmid M, Frey WH, Brünner YF, Chapman CD, Hölscher C, et al. Intranasal insulin as a treatment for Alzheimer's disease: a review of basic research and clinical evidence. CNS Drugs. 2013; 27: 505-514.
- Lieb W, Beiser AS, Vasan RS, Tan ZS, Au R, Harris TB, et al. Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA. 2009; 302: 2565-2572.
- Ishii M, Wang G, Racchumi G, Dyke JP, Ladecola C. Transgenic mice overexpressing amyloid precursor protein exhibit early metabolic deficits and a pathologically low leptin state associated with hypothalamic dysfunction in arcuate neuropeptide y neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014; 34: 9096-9106.
- Piacentini R, De Chiara G, Li Puma DD, Ripoli C, Marcocci ME, Garaci E, et al. HSV-1 and Alzheimer's disease: more than a hypothesis. Front Pharmacol. 2014; 5: 97.
- Itzhaki RF. Herpes simplex virus type 1 and Alzheimer's disease: increasing evidence for a major role of the virus. Front Aging Neurosci. 2014; 6: 202.
- Singhrao SK, Harding A, Simmons T, Robinson S, Kesavalu L, Crean S. Oral Inflammation, Tooth Loss, Risk Factors, and Association with Progression of Alzheimer's Disease. J Alzheimers Dis. 2014.
- Pisa D, Alonso R, Juarranz A, Rábano A, Carrasco L. Direct Visualization of Fungal Infection in Brains from Patients with Alzheimer's Disease. J Alzheimers Dis. 2014.
- De Chiara G, Marcocci ME, Sgarbanti R, Civitelli L, Ripoli C, Piacentini R, et al. Infectious agents and neurodegeneration. Mol Neurobiol. 2012; 46: 614-638.
- De Chiara G, Marcocci ME, Civitelli L, Argnani R, Piacentini R, Ripoli C, et al. APP processing induced by herpes simplex virus type 1 (HSV-1) yields several APP fragments in human and rat neuronal cells. PLoS One. 2010; 5: e13989.
- Piacentini R, Civitelli L, Ripoli C, Marcocci ME, De Chiara G, Garaci E, et al. HSV-1 promotes Ca2+ -mediated APP phosphorylation and Aβ accumulation in rat cortical neurons. Neurobiol Aging. 2011; 32: 2323.
- Wozniak MA, Itzhaki RF. Intravenous immunoglobulin reduces β amyloid and abnormal tau formation caused by herpes simplex virus type 1. J Neuroimmunol. 2013; 257: 7-12.
- 37. Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010; 5: e9505.
- Papareddy P, Mörgelin M, Walse B, Schmidtchen A, Malmsten M. Antimicrobial activity of peptides derived from humanß-amyloid precursor protein. J Pept Sci. 2012; 18: 183-191.
- White MR, Kandel R, Tripathi S, Condon D, Qi L, Taubenberger J, et al. Alzheimer's associatedß-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS One. 2014; 9: e101364.
- Hernández F, Gómez de Barreda E, Fuster-Matanzo A, Lucas JJ, Avila J. GSK3: a possible link between beta amyloid peptide and tau protein. Exp Neurol. 2010; 223: 322-325.
- Guo JP, Arai T, Miklossy J, McGeer PL. Abeta and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer's disease. Proc Natl Acad Sci U S A. 2006; 103: 1953-1958.
- Manczak M, Reddy PH. Abnormal interaction of oligomeric amyloid-β with phosphorylated tau: implications to synaptic dysfunction and neuronal damage. J Alzheimers Dis. 2013; 36: 285-295.
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, et al. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011; 31: 700-711.
- Minami SS, Clifford TG, Hoe HS, Matsuoka Y, Rebeck GW. Fyn knock-down increases Aβ, decreases phospho-tau, and worsens spatial learning in 3×Tg-AD mice. Neurobiol Aging. 2012; 33: 825.
- Cochran JN, Diggs PV, Nebane NM, Rasmussen L, White EL, Bostwick R, et al. AlphaScreen HTS and Live-Cell Bioluminescence Resonance Energy Transfer (BRET) Assays for Identification of Tau-Fyn SH3 Interaction Inhibitors for Alzheimer Disease. J Biomol Screen. 2014.
- Shukla V, Skuntz S, Pant HC. Deregulated Cdk5 activity is involved in inducing Alzheimer's disease. Arch Med Res. 2012; 43: 655-662.
- Piedrahita D, Hernández I, López-Tobón A, Fedorov D, Obara B, Manjunath BS, et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer's mice. J Neurosci. 2010; 30: 13966-13976.
- Zhang X, Hernandez I, Rei D, Mair W, Laha JK, Cornwell ME, et al. Diaminothiazoles modify Tau phosphorylation and improve the tauopathy in mouse models. J Biol Chem. 2013; 288: 22042-22056.
- van der Harg JM, Nölle A, Zwart R, Boerema AS, van Haastert ES, Strijkstra AM, et al. The unfolded protein response mediates reversible tau phosphorylation induced by metabolic stress. Cell Death Dis. 2014; 5: e1393.
- Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GVW. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J Biol Chem. 2009; 284: 18754-18766.
- Quintanilla RA, Dolan PJ, Jin YN, Johnson GV. Truncated tau and Aβ cooperatively impair mitochondria in primary neurons. Neurobiol Aging. 2012; 33: 619.
- Ramos-Rodriguez JJ, Molina-Gil S, Rey-Brea R, Berrocoso E, Alloza MG. Specific serotonergic denervation affects tau pathology and cognition without altering senile plaques deposition in APP/PS1 mice. PloS one. 2013; 8: e79947.
- Cohen SM. microRNAs in CNS Development and Neurodegeneration: Insights from Drosophila Genetics. Strooper Bd, Christen Y, editors. In: Macro Roles for MicroRNAs in the Life and Death of Neurons. Berlin: Springer-Verlag. 2010.
- Pham JT, Gallicano GI. Specification of neural cell fate and regulation of neural stem cell proliferation by microRNAs. Am J Stem Cells. 2012; 1: 182-195.
- Berezikov E, Thuemmler F, van Laake LW, Kondova I, Bontrop R, Cuppen E, et al. Diversity of microRNAs in human and chimpanzee brain. Nat Genet. 2006; 38: 1375-1377.
- Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, et al. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007; 317: 1220-1224.
- Wang G, van der Walt JM, Mayhew G, Li YJ, Züchner S, Scott WK, et al. Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of alpha-synuclein. Am J Hum Genet. 2008; 82: 283-289.
- Johnson R, Buckley NJ. Gene dysregulation in Huntington's disease: REST, microRNAs and beyond. Neuromolecular Med. 2009; 11: 183-199.
- Packer AN, Xing Y, Harper SQ, Jones L, Davidson BL. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington's disease. J Neurosci. 2008; 28: 14341-14346.
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003; 9: 3-4.
- Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer's disease hippocampus. Neuroreport. 2007; 18: 297-300.
- Sethi P, Lukiw WJ. Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer's disease temporal lobe neocortex. Neurosci Lett. 2009; 459: 100-104.
- Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, et al. The expression of microRNA miR-107 decreases early in Alzheimer's disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008; 28: 1213-1223.
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002; 59: 1381-1389.
- Sun A, Koelsch G, Tang J, Bing G. Localization of beta-secretase memapsin 2 in the brain of Alzheimer's patients and normal aged controls. Exp Neurol. 2002; 175: 10-22.
- Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor beta-secretase in Alzheimer's disease. Ann Neurol. 2002; 51: 783-786.
- Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T, et al. Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J Neurosci. 2007; 27: 3639-3649.
- Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, et al. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron. 2007; 54: 721-737.
- Hèbert SS, Horrè K, NicolaÏ L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008; 105: 6415-6420.
- Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, Cannon B, et al. Identification of miRNA changes in Alzheimer's disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis. 2008; 14: 27-41.
- Banzhaf-Strathmann J, Benito E, May S, Arzberger T, Tahirovic S, Kretzschmar H, et al. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer's disease. EMBO J. 2014; 33: 1667-1680.
- Zhao ZB, Wu L, Xiong R, Wang LL, Zhang B, Wang C, et al. MicroRNA-922 promotes tau phosphorylation by downregulating ubiquitin carboxy-terminal hydrolase L1 (UCHL1) expression in the pathogenesis of Alzheimer's disease. Neuroscience. 2014; 275: 232-237.
- Rodriguez-Ortiz CJ, Baglietto-Vargas D, Martinez-Coria H, LaFerla FM, Kitazawa M. Upregulation of miR-181 Decreases c-Fos and SIRT-1 in the Hippocampus of 3xTg-AD Mice. J Alzheimers Dis. 2014.
- Guedes JR, Custódia CM, Silva RJ, de Almeida LP, Pedroso de Lima MC, Cardoso AL. Early miR-155 upregulation contributes to neuroinflammation in Alzheimer's disease triple transgenic mouse model. Hum Mol Genet. 2014;.
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006; 15: 1437-1449.
- Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106: 14670-14675.
- Krako N, Magnifico MC, Arese M, Meli G, Forte E, Lecci A, et al. Characterization of mitochondrial dysfunction in the 7PA2 cell model of Alzheimer's disease. J Alzheimers Dis. 2013; 37: 747-758.
- Newington JT, Rappon T, Albers S, Wong DY, Rylett RJ, Cumming RC. Overexpression of pyruvate dehydrogenase kinase 1 and lactate dehydrogenase A in nerve cells confers resistance to amyloid beta and other toxins by decreasing mitochondrial respiration and reactive oxygen species production. J Biol Chem. 2012; 287: 37245-37258.
- Liu X, Xu K, Yan M, Wang Y, Zheng X. Protective effects of galantamine against Abeta-induced PC12 cell apoptosis by preventing mitochondrial dysfunction and endoplasmic reticulum stress. Neurochem Int. 2010; 57: 588-599.
- Wang K, Zhu L, Zhu X, Zhang K, Huang B, Zhang J, et al. Protective effect of paeoniflorin on Aβ25-35-induced SH-SY5Y cell injury by preventing mitochondrial dysfunction. Cell Mol Neurobiol. 2014; 34: 227-234.
- Reddy PH. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer's disease. Exp Neurol. 2009; 218: 286-292.
- Devi L, Ohno M. Mitochondrial dysfunction and accumulation of the β-secretase-cleaved C-terminal fragment of APP in Alzheimer's disease transgenic mice. Neurobiol Dis. 2012; 45: 417-424.
- Walls KC, Coskun P, Gallegos-Perez JL, Zadourian N, Freude K, Rasool S, et al. Swedish Alzheimer mutation induces mitochondrial dysfunction mediated by HSP60 mislocalization of amyloid precursor protein (APP) and beta-amyloid. J Biol Chem. 2012; 287: 30317-30327.
- Rhein V, Baysang G, Rao S, Meier F, Bonert A, Müller-Spahn F, et al. Amyloid-beta leads to impaired cellular respiration, energy production and mitochondrial electron chain complex activities in human neuroblastoma cells. Cell Mol Neurobiol. 2009; 29: 1063-1071.
- Manczak M, Reddy PH. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer's disease. Hum Mol Genet. 2012; 21: 5131-5146.
- Schuh RA, Jackson KC, Schlappal AE, Spangenburg EE, Ward CW, Park JH, et al. Mitochondrial oxygen consumption deficits in skeletal muscle isolated from an Alzheimer's disease-relevant murine model. BMC Neurosci. 2014; 15: 24.
- Hroudová J, Singh N, Fišar Z. Mitochondrial dysfunctions in neurodegenerative diseases: relevance to Alzheimer's disease. Biomed Res Int. 2014; 2014: 175062.
- Shaerzadeh F, Motamedi F, Khodagholi F. Inhibition of Akt Phosphorylation Diminishes Mitochondrial Biogenesis Regulators, Tricarboxylic Acid Cycle Activity and Exacerbates Recognition Memory Deficit in Rat Model of Alzheimer's Disease. Cell Mol Neurobiol. 2014.
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004; 304: 448-452.
- Endres K, Reinhardt S. ER-stress in Alzheimer's disease: turning the scale? Am J Neurodegener Dis. 2013; 2: 247-265.
- Mattson MP, Guo Q. Cell and molecular neurobiology of presenilins: a role for the endoplasmic reticulum in the pathogenesis of Alzheimer's disease? J Neurosci Res. 1997; 50: 505-513.
- Katayama T, Imaizumi K, Honda A, Yoneda T, Kudo T, Takeda M, et al. Disturbed activation of endoplasmic reticulum stress transducers by familial Alzheimer's disease-linked presenilin-1 mutations. J Biol Chem. 2001; 276: 43446-43454.
- Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M. Induction of neuronal death by ER stress in Alzheimer's disease. J Chem Neuroanat. 2004; 28: 67-78.
- Brown MK, Naidoo N. The endoplasmic reticulum stress response in aging and age-related diseases. Front Physiol. 2012; 3: 263.
- Barbero-Camps E, Fernández A, Baulies A, Martinez L, Fernández-Checa JC, Colell A. Endoplasmic reticulum stress mediates amyloid-ß neurotoxicity via mitochondrial cholesterol trafficking. Am J Pathol. 2014; 184: 2066-2081.
- Fonseca AC, Ferreiro E, Oliveira CR, Cardoso SM, Pereira CF. Activation of the endoplasmic reticulum stress response by the amyloid-beta 1-40 peptide in brain endothelial cells. Biochim Biophys Acta. 2013; 1832: 2191-2203.
- Reinhardt S, Schuck F, Grösgen S, Riemenschneider M, Hartmann T, Postina R, et al. Unfolded protein response signaling by transcription factor XBP-1 regulates ADAM10 and is affected in Alzheimer's disease. FASEB J. 2014; 28: 978-997.
- Nijholt DA, van Haastert ES, Rozemuller AJ, Scheper W, Hoozemans JJ. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J Pathol. 2012; 226: 693-702.
- Abisambra JF, Jinwal UK, Blair LJ, O'Leary JC 3rd, Li Q, Brady S, et al. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J Neurosci. 2013; 33: 9498-9507.
- Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012; 2: a006346.
- Persson T, Popescu BO, Cedazo-Minguez A. Oxidative stress in Alzheimer's disease: why did antioxidant therapy fail? Oxid Med Cell Longev. 2014; 2014: 427318.
- Enciu AM, Popescu BO. Is there a causal link between inflammation and dementia? Biomed Res Int. 2013; 2013: 316495.
- Apelt J, Schliebs R. Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 2001; 894: 21-30.
- Tooyama I, Kimura H, Akiyama H, McGeer PL. Reactive microglia express class I and class II major histocompatibility complex antigens in Alzheimer's disease. Brain Res. 1990; 523: 273-280.
- Perlmutter LS, Scott SA, Barrón E, Chui HC. MHC class II-positive microglia in human brain: association with Alzheimer lesions. J Neurosci Res. 1992; 33: 549-558.
- Uchihara T, Akiyama H, Kondo H, Ikeda K. Activated microglial cells are colocalized with perivascular deposits of amyloid-beta protein in Alzheimer's disease brain. Stroke. 1997; 28: 1948-1950.
- Vehmas AK, Kawas CH, Stewart WF, Troncoso JC. Immune reactive cells in senile plaques and cognitive decline in Alzheimer's disease. Neurobiol Aging. 2003; 24: 321-331.
- Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006; 3: 27.
- Mizuno T. The biphasic role of microglia in Alzheimer's disease. Int J Alzheimers Dis. 2012; 2012: 737846.
- Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003; 23: 2665-2674.
- Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, et al. Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nat Med. 2007; 13: 1359-1362.
- Gómez Ravetti M, Moscato P. Identification of a 5-protein biomarker molecular signature for predicting Alzheimer's disease. PLoS One. 2008; 3: e3111.
- Christen Y. Oxidative stress and Alzheimer disease. Am J Clin Nutr. 2000; 71: 621S-629S.
- Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer's disease. Ann N Y Acad Sci. 2008; 1147: 180-195.
- Atwood CS, Perry G, Zeng H, Kato Y, Jones WD, Ling KQ, et al. Copper mediates dityrosine cross-linking of Alzheimer's amyloid-beta. Biochemistry. 2004; 43: 560-568.
- De Pittá M, Volman V, Berry H, Parpura V, Volterra A, Ben-Jacob E. Computational quest for understanding the role of astrocyte signaling in synaptic transmission and plasticity. Front Comput Neurosci. 2012; 6: 98.
- Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, et al. Astrocyte senescence as a component of Alzheimer's disease. PLoS One. 2012; 7: e45069.
- Chow SK, Yu D, Macdonald CL, Buibas M, Silva GA. Amyloid beta-peptide directly induces spontaneous calcium transients, delayed intercellular calcium waves and gliosis in rat cortical astrocytes. ASN Neuro. 2010; 2: e00026.
- Mei X, Ezan P, Giaume C, Koulakoff A. Astroglial connexin immunoreactivity is specifically altered atβ- amyloid plaques in β- amyloid precursor protein/presenilin1 mice. Neuroscience. 2010; 171: 92-105.
- Mulder SD, Veerhuis R, Blankenstein MA, Nielsen HM. The effect of amyloid associated proteins on the expression of genes involved in amyloid-β clearance by adult human astrocytes. Exp Neurol. 2012; 233: 373-379.
- Skaper SD, Evans NA, Rosin C, Facci L, Richardson JC. Oligodendrocytes are a novel source of amyloid peptide generation. Neurochem Res. 2009; 34: 2243-2250.
- Zhao J, O'Connor T, Vassar R. The contribution of activated astrocytes to Aß production: implications for Alzheimer's disease pathogenesis. J Neuroinflammation. 2011; 8: 150.
- Glenner GG. Congophilic microangiopathy in the pathogenesis of Alzheimer's syndrome (presenile dementia). Med Hypotheses. 1979; 5: 1231-1236.
- Zlokovic BV. Clearing amyloid through the blood-brain barrier. J Neurochem. 2004; 89: 807-811.
- Pluta R, Amek MU. Brain ischemia and ischemic blood-brain barrier as etiological factors in sporadic Alzheimer's disease. Neuropsychiatr Dis Treat. 2008; 4: 855-864.
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000; 106: 1489-1499.
- Jeynes B, Provias J. Significant negative correlations between capillary expressed eNOS and Alzheimer lesion burden. Neurosci Lett. 2009; 463: 244-248.
- Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, Sharom FJ, et al. beta-Amyloid efflux mediated by p-glycoprotein. J Neurochem. 2001; 76: 1121-1128.
- Bartels AL, Willemsen AT, Kortekaas R, de Jong BM, de Vries R, de Klerk O, et al. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson's disease, PSP and MSA. J Neural Transm. 2008; 115: 1001-1009.
- Qosa H, Abuasal BS, Romero IA, Weksler B, Couraud PO, Keller JN, et al. Differences in amyloid-β clearance across mouse and human blood-brain barrier models: kinetic analysis and mechanistic modeling. Neuropharmacology. 2014; 79: 668-678.
- Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. 2009; 118: 103-113.
- Nakajima M, Yuasa S, Ueno M, Takakura N, Koseki H, Shirasawa T. Abnormal blood vessel development in mice lacking presenilin-1. Mech Dev. 2003; 120: 657-667.
- Meyer EP, Ulmann-Schuler A, Staufenbiel M, Krucker T. Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer's disease. Proc Natl Acad Sci U S A. 2008; 105: 3587-3592.
- Desai BS, Schneider JA, Li JL, Carvey PM, Hendey B. Evidence of angiogenic vessels in Alzheimer's disease. J Neural Transm. 2009; 116: 587-597.
- Gheorghiu M, Enciu AM, Popescu BO, Gheorghiu E. Functional and molecular characterization of the effect of amyloid-β 42 on an in vitro epithelial barrier model. J Alzheimers Dis. 2014; 38: 787-798.
- Qosa H, LeVine H, 3rd, Keller JN, kaddoumi A. Mixed oligomers and monomeric amyloid-beta disrupts endothelial cells integrity and reduces monomeric amyloid-beta transport across hCMEC/D3 cell line as an in vitro blood-brain barrier model. Biochim Biophys Acta. 2014; 1842: 1806-1815.
- Lovell MA, Geiger H, Van Zant GE, Lynn BC, Markesbery WR. Isolation of neural precursor cells from Alzheimer's disease and aged control postmortem brain. Neurobiol Aging. 2006; 27: 909-917.
- Zhang C, McNeil E, Dressler L, Siman R. Long-lasting impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer's disease. Experimental neurology. 2007; 204: 77-87.
- Li B, Yamamori H, Tatebayashi Y, Shafit-Zagardo B, Tanimukai H, Chen S, et al. Failure of neuronal maturation in Alzheimer disease dentate gyrus. J Neuropathol Exp Neurol. 2008; 67: 78-84.
- Mazur-Kolecka B, Golabek A, Nowicki K, Flory M, Frackowiak J. Amyloid-beta impairs development of neuronal progenitor cells by oxidative mechanisms. Neurobiol Aging. 2006; 27: 1181-1192.
- Calafiore M, Battaglia G, Zappalà A, Trovato-Salinaro E, Caraci F, Caruso M, et al. Progenitor cells from the adult mouse brain acquire a neuronal phenotype in response to beta-amyloid. Neurobiol Aging. 2006; 27: 606-613.
- Chen Y, Dong C. Abeta40 promotes neuronal cell fate in neural progenitor cells. Cell Death Differ. 2009; 16: 386-394.
- Sotthibundhu A, Li QX, Thangnipon W, Coulson EJ. Abeta(1-42) stimulates adult SVZ neurogenesis through the p75 neurotrophin receptor. Neurobiol Aging. 2009; 30: 1975-1985.
- Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP. Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer's disease. J Neurochem. 2002; 83: 1509-1524.
- Chuang TT. Neurogenesis in mouse models of Alzheimer's disease. Biochim Biophys Acta. 2010; 1802: 872-880.
- Kolecki R, Lafauci G, Rubenstein R, Mazur-Kolecka B, Kaczmarski W, Frackowiak J. The effect of amyloidosis-beta and ageing on proliferation of neuronal progenitor cells in APP-transgenic mouse hippocampus and in culture. Acta Neuropathol. 2008; 116: 419-424.
- He N, Jin WL, Lok KH, Wang Y, Yin M, Wang ZJ. Amyloid-β(1-42) oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis. 2013; 4: e924.
- Lee IS, Jung K, Kim IS, Park KI. Amyloid-β oligomers regulate the properties of human neural stem cells through GSK-3ß signaling. Exp Mol Med. 2013; 45: e60.
- Leonard BW, Mastroeni D, Grover A, Liu Q, Yang K, Gao M, et al. Subventricular zone neural progenitors from rapid brain autopsies of elderly subjects with and without neurodegenerative disease. The Journal of comparative neurology. 2009; 515: 269-294.
- Kronenberg H. Adult mesenchymal stem cells. StemBook. 2009.
- Ozen I, Boix J, Paul G. Perivascular mesenchymal stem cells in the adult human brain: a future target for neuroregeneration? Clin Transl Med. 2012; 1: 30.
- Lee JK, Jin HK, Endo S, Schuchman EH, Carter JE, Bae JS. Intracerebral transplantation of bone marrow-derived mesenchymal stem cells reduces amyloid-beta deposition and rescues memory deficits in Alzheimer's disease mice by modulation of immune responses. Stem Cells. 2010; 28: 329-343.
- Ma T, Gong K, Ao Q, Yan Y, Song B, Huang H, et al. Intracerebral transplantation of adipose-derived mesenchymal stem cells alternatively activates microglia and ameliorates neuropathological deficits in Alzheimer's disease mice. Cell transplantation. 2013; 22: S113-126.