
Abstract
Rosai-Dorfman Disease (RDD), is a rare and self-limiting benign proliferation of histiocytes with unknown etiologies. It is also known as Sinus Histiocytosis with Massive Lymphadenopathy (SHML), which was first reported by Rosai and Dorfman in 1969. The disease is classified as an idiopathic non-Langerhans cell histiocytosis which most commonly involves the cervical lymph nodes [1]. It is clinically characterized as massive, painless, bilateral and symmetric cervical lymphadenopathy accompanied with fever and leukocytosis. In the past several decades, there have been a few studies on the skin, respiratory tract, bone, genitourinary system, oral cavity, central nervous system, eyes/orbit/ocular adnexa, salivary gland, tonsil, breast, soft tissue, and heart.
Confirming the diagnosis of RDD remains challenging, despite improvements in both histological and imaging techniques. In most cases, the diagnoses are made postoperatively based on the histological findings.
In this report, a unique case about a 63-year-old female who was diagnosed as ulnar RDD was presented. We describe the details of the diagnosis and treatment of the patient and provide a review of the literature.
Keywords: Immunohistochemistry ulna; Rosai-Dorfman disease: Elbow; Extranodal Sinus Histiocytosis with Massive Lymphadenopathy (SHML)
Case Report
A 63-year-old female was admitted to the hospital with a twomonth history of dull pain in the right elbow. Her arm tingled, but no numbness or any radiation of pain down the affected arm. Her pain was serious for hyperactivity and lifting heavy weights. Clinical examination showed there was some swelling and edema in the right elbow. Surrounding the edema is most likely to be a result of reaction to the osseous lesions because it can also be seen in many other bone lesions, and no overlying skin erythema. Cervical, axillary and inguinal lymphadenopathies were not appreciated. Ulnar side elbow had obvious local tenderness and no abnormality, superficial varicose and activity limitation. The patient denied any fever, medical problems or drug allergies. The previous history of lymph node enlargement was denied by the patient.
The haemoglobin was 123g/L (normal 113-151 g/L), white cell count was 6300/mm3 (normal 3690-9160/mm3) and the Erythrocyte Sedimentation Rate (ESR) was 21mm/h (normal 0-20 mm/h). The albumin, globulin, alkaline phosphatase, serum calcium and phosphate levels were within normal limits. Quantiferon T-spot TB test is negative. Antinuclear Antibodies (ANA), Antineutrophil Cytoplasmic Antibody (ANCA), EBV, Epstein-Barr Virus (EBV) and herpes virus is negative. Chest radiographs were normal. Plain radiographs of the right elbow revealed that right proximal ulna destruction were irregular because a little periosteal reaction side of the rim no obvious narrow joint space (Figure 1A). The patient was referred for a Magnetic Resonance Imaging (MRI) to further study the elbow pain. The MRI showed mild expansile destructive lesion in right proximal ulna (including the coronoid process) and abnormal signal was about 5.6cm×1.9cm×2.2cm without clear border to the medullary cavity. The lesion appeared to be slight hypo- or hypo-intensity in T1-weighted images and slightly hyper- or hyperintensity in T2-weighted images. Images enhanced markedly after contrast-enhanced scanning. The lesion extended beyond the cortical margins of the bone and went into the adjacent muscle (Figure 1B).
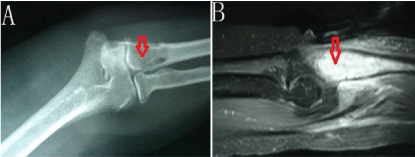
Figure 1: A) Plain radiographs of the right elbow revealed that right proximal
ulna destruction were irregular because a little periosteal reaction side
of the rim no obvious narrow joint space. B) Images enhanced markedly
after contrast-enhanced scanning. The lesion extended beyond the cortical
margins of the bone and went into the adjacent muscle.
Then the patient underwent a computed tomography guided biopsy of the lesion. The histology showed granulation with many inflammatory cell infiltrations. As the cytologic diagnosis was not reflective of the radiologic or clinical impression, 5 days later, the woman underwent the lesion removal in the right ulna. Frozen pathological section showed caseous institution in the medullary cavity and the right elbow joints, bone and soft tissue of chronic inflammation associated with inflammatory granulation tissue formation and foamy cell reaction. The histiocytes staining showed positive S-100 and CD68, negative CD1a (Figure 2). The histologic appearance of the lesion and the immunohistochemical findings were consistent with a diagnosis of RDD.
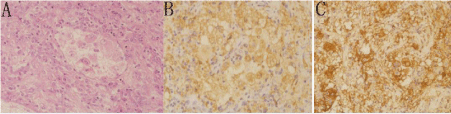
Figure 2: The histiocytes staining showed positive S-100 and CD68, negative
CD1a.
Three-Year Follow-Up
The patient did not receive any treatment after the surgery. By the start of the following study (three years after the surgery), the patient had regained full strength, velocity comparing to the situation before the surgery. At the latest follow-up (after 3 years) the patient remained asymptomatic and normal magnetic resonance imaging. It demonstrated an intact ulna without evidence of recurrence.
Discussion
RDD is a rare, benign histiocytosis and is most commonly characterized as painless, massive cervical lymphadenopathy which affects all age groups including pediatric patients [2]. There is a slight predilection in male and African Americans that they are more likely to be affected. The commonest presentation of RDD is painless cervical lymphadenopathy. However, other nodal sites may be involved. In the order of frequency, the sites are cervical, axillary, inguinal, paraaortic, and mediastinal lymph nodes. Approximately 90% of patients will have painless lymphadenopathy in the cervical region. 43% of the patients suffer from extra-nodal disease and only 23% suffer from extra-nodal disease without identified nodal involvement [3]. Rare extranodal sites include the soft tissue, skin, upper respiratory tract, gastrointestinal tract, breast, bones, and the central nervous system. In the 423 RDD cases presented by Foucar et al, 7.2% of cases had osseous involvement without lymphadenopathy, and 0.5% of them had isolated osseous involvement [3]. However, constitutional signs and symptoms might be evidences, such as fever, weight loss, leukocytosis and neutrophilia. Signs and symptoms of the disease are usually various according to the affected sites. RDD is easily misdiagnosed due to the clinical symptoms, rarity, and lack of understanding of this disease. The clinical symptom of the case reported here was some pain in right elbow without any fever and other medical problems.
Although the case of intrasseous RRD without identified nodal involvement is rare, it has been already reported. In a recent case, series of primary intraosseous RDD lesions were found in the tibia, femur, clavicle, skull, maxilla, calcaneus, phalanx, metacarpal, glenoid, mandible, carpal, triquetrum and sacrum [4-6]. The average age of presentation was 27 years old [4]. A recent case report by Bachmann et al also found osseous RDD in the ileum in a 71-yearold patient, which is far beyond the typical age of presentation [7]. Compared with its presentation in lymph nodes, osseous RDD has less pronounced lymphophagocytosis and more fibrosis.
RDD is a disorder characterized by non-malignant proliferation of distinctive histiocytic/phagocytic cells primarily within lymph node sinuses and lymphatics in extranodal sites. The differential diagnosis includes Langerhans Cell Histiocytosis (LCH), osteomyelitis, lymphoma, metastatic carcinomatous deposits, and infective tenosynovitis (nonspecific and tubercular). RDD shows dilated sinuses filled with lymphocytes and numerous histiocytes with intact lymphocytes within their cytoplasm, a feature known as emperipolesis or lymphophagocytosis. The germinal centers may be hyperplastic, or even sparse to absent. LCH is a dendritic cell proliferative disorder, while RDD is a monocyte/macrophage proliferative disease. Osteomyelitis usually shows chronic and acute inflammatory cells in a necrotic background. Infective synovitis contains synoviocytes, acute and chronic inflammatory cells. In lymphoma, the above features are inconspicuous. Metastasis is ruled out by absence of malignant epithelial cells. Tubercular synovitis contains tubercular granuloma. In order to rule out bone tuberculosis, T-spot IGFA was specially done for this patients reported.
Laboratory tests showed no specific abnormalities in RDD. The diagnosis was made on the basis of characteristic clinical features (particularly the presence of painless adenopathy with or without skin or other manifestations) combined with characteristic histologic findings in tissue biopsy specimens. The patient in the current report exhibited only localized elbow pain and swelling, while did not demonstrate any other signs or symptoms. The diagnosis can be as difficult as the patient may not have the classic features associated with nodal disease such as fever, leukocytosis, and elevated erythrocyte sedimentation rate.
The main features on histology are characteristic pattern of cell surface marker expressions: positive expression of S100 protein, CD14, CD68 (macrophage markers), positive for CD11c (early dendritic cell marker), but negative for CD1a (positive on Langerhanstype of dendritic cells) and MHC-2 (generally positive on dendritic cells). Thus the cell of origin of RDD is presumed to be a monocyte-derived cell that is on its way to macrophage differentiation and at the same time has some markers of early dendritic cell phenotype.
Though RDD is a self-limiting disorder, the course of RDD can sometimes behave in an aggressive manner, leading to significant morbidity and mortality because of organ involvement or systemic manifestations. In most cases localized extranodal disease, a primarily surgical therapy was prompted to accept. In several cases antibiotics or tuberculostatic agents were given without a significant response. Corticosteroids have been tried successfully in generalized lymphadenopathy or in a case with intracranial infiltrations [8]. Treatment with chemotherapeutics may be indicated and have been tried by Chu P and Foucar E [9,10]. However, four patients died despite of chemotherapy [10]. Interferon-a (IFN) has been previously reported in only one case and the result is effective. More experience is needed to evaluate the role of IFN [11].
References
- Vermeulen TL, Isaacs TW, Spagnolo D, Amanuel B. Rosai-Dorfman disease presenting as choroidal melanoma: a case report and review of the literature. Arch Clin Exp Ophthalmol. 2013; 251: 295-299.
- Ku PK, Tong MC, Leung CY, Pak MW, van Hasselt CA. Nasal manifestation of extranodal Rosai-Dorfman disease-diagnosis and management. J Laryngol Otol. 1999; 113: 275-280.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990; 7: 19-73.
- Tripathy K, Misra A, Sahu AK, Patnaik K. Extranodal Rosai-Dorfman disease in a carpal bone. Indian J Orthop. 2012; 46: 487-489.
- Demicco EG, Rosenberg AE, Bjornsson J, Rybak LD, Unni KK, Nielsen GP. Primary Rosai-Dorfman disease of bone: a clinicopathologic study of 15 cases. Am J Surg Pathol. 2010; 34: 1324-1333.
- Hsu A, Bhatia S, Kang RW, Arvanitis L, Nicholson GP, Virkus WW. Extranodal Rosai-Dorfman disease presenting as an isolated glenoid lesion in a high school athlete. J Shoulder Elbow Surg. 2012; 21: 6-11.
- Bachmann K, Dragoescu E, Foster W. Extranodal Rosai-Dorfman disease presenting as incidental bone tumor: a case report. Am J Orthop. 2010; 39: E123-E125.
- Shaver EG, Rebsamen SL, Yachnis AT, Sutton LN. Isolated extranodal intracranial sinus histiocytosis in a 5-year-old boy. J Neurosurg. 1993; 79: 769-773.
- Chu P, LeBoit PE. Histologic features of cutaneous sinus histiocytosis (Rosaie Dorfman disease): study of cases both with and without systemic involvement. J Cutan Pathol. 1992; 19: 201-206.
- Foucar E, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: An analysis of 14 deaths occurring in a patients registry. Cancer. 1984; 54: 1834-1840.
- Pulsoni A, Anghel G, Falcucci P, Matera R, Pescarmona E, Ribersani M, et al. Treatment of sinus histiocytosis with massive lymphadenopathy (RosaieDorfman disease): report of a case and literature review. Am J Hematol. 2002; 69: 67-71.