
Abstract
This study reports an outbreak of Tuberculosis (TB) in a high school in Hunan province, China during January 2017-April 2018. Contact investigation and TB screening were conducted through symptom screening, tuberculin skin testing, chest radiography and smear examination. Identification of positive isolates and drug susceptibility phenotype were assessed by standard method. Mycobacterial Interspersed Repetitive Units-Variable Number Tandem Repeats (MIRU-VNTR) and Whole Genome Sequencing (WGS) were performed to investigate the relationships among the positive isolates. A total of 90 students and one teacher were diagnosed active pulmonary TB among 2908 students and 188 staff, with an attack rate of 2.94%. Thirteen positive isolates were identified as drug susceptible Beijing family of Mycobacterium tuberculosis. Results of MIRU-VNTR typing and WGS revealed two clones of Mycobacterium tuberculosis circulating during outbreak. One hundred and twenty-nine Single Nucleotide Polymorphisms (SNPs) discriminated the isolates in two clusters; the maximum number of SNPs between any pair of isolates in each cluster was five or fewer. Our findings highlight the importance of early identification and isolation of the TB cases to prevent spread of TB. WGS provides better resolution than MIRU-VNTR to identify recent transmission in TB outbreak.
Keywords: Tuberculosis; School; Outbreak; MIRU-VNTR typing; Whole genome sequencing
Introduction
China is a country with high burden of Tuberculosis (TB) and drug resistant TB. According to the latest report of the World Health Organization (WHO), the number of TB in China ranked the third place in the world [1]. Current measures to prevent and control of TB are often focused on adults, while adolescents are often neglected in China. In recent years, some school based TB outbreaks have been reported in China [2-7], which attracts considerable attention. Data from released annual report on surveillance of infectious disease in China in 2017, 835193 TB cases were notified in China, among which, 40656 (4.87%) were students, incidence of pulmonary TB was 15.47/100000 students, and incidence of smear positive of pulmonary TB was 2.64/100000 students, the proportion of laboratory confirmed TB ranged from 8.3% to 20.8% [8]. In China, high school students face serious challenge for college entrance examination, because of high pressure and overcrowded conditions, school based TB outbreaks usually occurred in boarding school system [2,7]. Diagnosis of tuberculosis in students often relies on symptoms screening, chest radiography and smear examination, and the low proportion of laboratory confirmed TB and few of positive strains isolated from TB students, which make transmission investigation more difficult.
In the current study, we used traditional epidemiology investigation combined with Mycobacterial Interspersed Repetitive Units-Variable Number Tandem Repeats (MIRU-VNTR) genotyping and Whole Genome Sequencing (WGS) to delineate a high school TB outbreak.
Methods
Study setting
This TB outbreak was in a high school in Hunan province. Hunan province locates in central area of China with 68.6 million inhabitants, the reported incidence of TB was 76.07/100000 people in 2017. The high school is located in a county with 0.89 million inhabitants. TB cases are diagnosed and treated by local health service system.
Founded in 1956, this high school covers an area of 7.2 hectares. It has three teaching buildings, three teaching staff accommodation buildings, three student dormitory buildings, a science and technology building, a track and field ground and a canteen. During TB outbreak, this school had 188 staff, 2908 students were attending school constituting three grades from senior one to three with 852, 1023 and 1033 students respectively. Classroom is about 50 m2, class A senior three with the largest number of TB cases had 99 students in the classroom, four air conditioners and six fans were used for temperature adjustment, and windows were seldom opened for ventilation. Student’s dormitory is about 20 m² with 14 beds, and each dormitory equipped an air conditioner has a separate toilet, ventilation conditions are poor. Because of overcrowded dormitory, most students rented houses near the school and lived with their parents.
Epidemiological investigation and TB screening
Contact investigation was conducted in accordance with Chinese guidelines for TB prevention and control in school, contacts were suggested to do symptoms screening first, and then Tuberculin Skin Test (TST), positive TST (induration of 5 mm or larger) contacts were recommended to perform radiography or Computed Tomography (CT) and smear examination. Contacts investigation was performed in all students, faculty members, canteen staff, administrative staff and family members of TB students. Students and staff with positive TST underwent clinical evaluation, chest radiography and smear examination evaluation; they were defined as latent TB infection if diagnosis of active TB was ruled out.
Smear, culture, identification, and drug susceptibility testing of Mycobacterium tuberculosis (M. tuberculosis)
Specimens were collected from suspected TB cases, microscopy and culture of mycobacteria on Löwenstein-Jensen (L-J) medium were performed using standard methods at local TB laboratory, and sputum specimens of presumptive TB students were tested by Xpert MTB/RIF assay according to manufacturer’s instructions at provincial TB laboratory. Acid fast bacilli positive isolates were sent to the national TB reference laboratory, identification of mycobacterium was performed by testing isolates susceptibility to p-nitrobenzoic acid, and drug susceptibility to rifampin, isoniazid, streptomycin, ethambutol, kanamycin and ofloxacin was performed using proportional method on L-J medium according to WHO standard method.
Identification of M. tuberculosis Beijing family
M. tuberculosis Beijing family was identified via RD-105 multiplex PCR [9]. Modern and ancient Beijing families were identified by polymorphism of the NTF locus as previously study reported [10].
MIRU-VNTR genotyping and WGS
To determine a detailed profile of TB outbreak, a standardized set of 24-loci MIRU-VNTR genotyping was performed as previously study reported [11]. Two or more isolates with indistinguishable MIRU-VNTR genotypes were assigned to cluster.
DNA was extracted from positive isolates using standardized procedures. Sequencing libraries were prepared from 100ng DNA using the TruSeq sample preparation kit (Illumina Inc.) according to the manufacturer’s instructions guide. Genomic DNA was sequenced on the HiSeq X Ten System, over 100× coverage could be achieved with 6 million reads of 150 bp paired-end reads. Valid reads were aligned to the H37Rv reference genome (GenBank accession no. NC_000962.3) by using Burrows-Wheeler aligner v0.71 (http://biobwa. sourceforge.net/) [12], single nucleotide polymorphisms (SNPs) were identified by Speedseq [13], variations were annotated by SnpEff [14].
SNPs located in PE/PPE/PGRS genes, repetitive regions and transposons were excluded from the analyses to avoid possible errors in the read alignment in those regions of the genome. Furthermore, SNPs in drug resistance associated genes were also removed to avoid the possible influence on the reliability of phylogeny [15]. The concatenated alignment was used to generate phylogenetic tree, MEGA X software was employed to create a Maximum Likelihood tree for positive isolates [16].
Ethical approval
According to Chinese national TB control program, this study was a response to public emergency event, ethical approval was not required to investigate and report on infectious disease outbreak.
Results
TB outbreak in high school
In January 2017, a high school student in class A senior three developed cough and went to outpatient department of local center for disease control and prevention (CDC), the student was diagnosed smear positive TB (acid fast bacilli 1+), because of concealment of his student identity, the doctor treated this case as sporadic case. In the following months of February, April, May and June of 2017, four students from this class were diagnosed pulmonary TB one after another, but none of these cases told doctors their students’ identity. On July 26, 2017, three students came to the county CDC, one suspected TB for diagnosis, two TB cases for treatment follow up visit, from their conversation, the doctor found out they knew each other and suspected they were students from same school. After detailed inquiry, three cases admitted they were from same high school, which alerted the CDC staff, epidemiology contact investigation was conducted promptly. Contacts were screened by symptoms, TST, radiography or CT and smear examination, 91 cases including 90 students and one teacher were diagnosed active TB by the end of 30 April 2018, with an attack rate of 2.94%, all TB cases were subjected to standard treatment. Fifty-seven students with strong positive TST (induration of 15 mm or larger) were defined as latent TB infection and recommended taking preventive treatment.
Ninety students were diagnosed pulmonary TB in this outbreak, among them, seven and 83 students were in senior two and three grade, with attack rate 0.68% and 8.03% respectively. In senior three, 67.47% (56/83) of the TB students were in class A, the attack rate of the students in class A was 56.56%.
Among 91 confirmed TB cases, acid fast bacilli positive strains were isolated from 13 cases, including two male and 11 female students with median age 17 years (16 to 19 years). Nine TB cases were in class A, the remaining were scattered over four classes but in same grade, the characteristics of 13 culture positive cases were showed in Table 1.
![]()
Case
Grade
Class
Age (Y)
Sex
Smear
TST
CT
Xpert MTB/RIF
Date of diagnosis
1
Three
A
19
Female
Negative
Strong positive
Tuberculosis in the right lower lung
Not done
7-Nov-17
2
Three
A
17
Female
Positive
Strong positive
Bilateral pulmonary tuberculosis
M.TB
RIF sensitive20-Aug-17
3
Three
A
17
Female
Negative
Strong positive
Tuberculosis in the left upper lung
Not done
17-Aug-17
4
Three
A
16
Female
Negative
Strong positive
Bilateral pulmonary tuberculosis
Not done
19-Aug-17
5
Three
B
17
Male
Negative
Strong positive
Tuberculosis in the right upper lung
Negative
29-Aug-17
6
Three
C
17
Female
Negative
Strong positive
Tuberculosis in the left upper lung
Negative
22-Aug-17
7
Three
A
17
Female
Negative
Strong positive
Tuberculosis in the left lower lung
Not done
12-Nov-17
8
Three
D
18
Female
Not done
Strong positive
Not available
Not done
21-Aug-17
9
Three
A
17
Female
Positive
Negative
Tuberculosis in the right upper and left lower lung
M.TB
RIF sensitive7-Nov-17
10
Three
A
18
Male
Positive
Not done
Tuberculosis in the left upper and middle lung
Not done
8-Aug-17
11
Three
E
18
Female
Negative
Strong positive
Tuberculosis in the left upper lung
Negative
23-Aug-17
12
Three
A
17
Female
Positive
Not done
Bilateral pulmonary tuberculosis
Not done
19-Aug-17
13
Three
A
18
Female
Not available
Not available
Not available
Not available
Not available
CT:Computerized Tomography; M.TB: Mycobacterium Tuberculosis; RIF: Rifampin
Table 1: Characteristics of the culture positive active tuberculosis cases identified during high school outbreak, China.
Characteristics of outbreak isolates
Thirteen acid fast bacilli positive isolates were identified as M. tuberculosis. The results of drug susceptibility testing showed that all isolates were susceptible to rifampin, isoniazid, ethambutol, streptomycin, kanamycin and ofloxacin. All isolates were identified Beijing family, among which, two (15%) isolates (No.2 and No.8) were identified modern Beijing family; the remaining 11 (85%) isolates were identified ancient Beijing family. Sequencing of drug resistance conferring genes including rpoB, inhA, KatG, rrS, embB, gyrA, eis and pncA showed that no mutation in these genes were identified.
Outbreak defined by MIRU-VNTR and WGS
MIRU-VNTR and WGS were performed on the 13 M. tuberculosis isolates. Results of 24-loci MIRU-VNTR typing showed that six loci variations discriminated the isolates into two clusters, the major cluster consisted of 11 ancient Beijing family isolates, the smaller cluster was comprised of two modern Beijing family isolates (No. 2 and No.8), the two clusters differed by variations at locus MIRU26, MIRU10, QUB-4156, QUB-11b, Mtub21 and QUB-26 (Table 2), the results of MIRU-VNTR suggested two clonal outbreaks.
![]()
ID
MIRU04
MIRU 26
MIRU 40
MIRU 10
MIRU 31
MIRU 16
Mtub04
ETR A
ETRC
Mtub30
Mtub39
QUB-4156
QUB-11b
Mtub21
QUB-26
MIRU02
MIRU23
MIRU39
MIRU20
MIRU24
MIRU27
Mtub29
ETR-B
Mtub34
1
3
8
3
3
3
5
4
4
4
4
2
4
7
4
7
2
5
3
2
1
3
4
2
3
2
3
7
3
2
3
5
4
4
4
4
2
2
6
5
8
2
5
3
2
1
3
4
2
3
3
3
8
3
3
3
5
4
4
4
4
2
4
7
4
7
2
5
3
2
1
3
4
2
3
4
3
8
3
3
3
5
4
4
4
4
2
4
7
4
7
2
5
3
2
1
3
4
2
3
5
3
8
3
3
3
5
4
4
4
4
2
4
7
4
7
2
5
3
2
1
3
4
2
3
6
3
8
3
3
3
5
4
4
4
4
2
4
7
4
7
2
5
3
2
1
3
4
2
3
7
3
8
3
3
3
5
4
4
4
4
2
4
7
4
7
2
5
3
2
1
3
4
2
3
8
3
7
3
2
3
5
4
4
4
4
2
2
6
5
8
2
5
3
2
1
3
4
2
3
9
3
8
3
3
3
5
4
4
4
4
2
4
7
4
7
2
5
3
2
1
3
4
2
3
10
3
8
3
3
3
5
4
4
4
4
2
4
7
4
7
2
5
3
2
1
3
4
2
3
11
3
8
3
3
3
5
4
4
4
4
2
4
7
4
7
2
5
3
2
1
3
4
2
3
12
3
8
3
3
3
5
4
4
4
4
2
4
7
4
7
2
5
3
2
1
3
4
2
3
13
3
8
3
3
3
5
4
4
4
4
2
4
7
4
7
2
5
3
2
1
3
4
2
3
MIRU-VNTR: Mycobacterial Interspersed Repetitive Units-Variable Number Tandem Repeats.
Table 2: Profiles of MIRU-VNTR genotyping of M. tuberculosis isolated from culture positive active tuberculosis cases during high school TB outbreak.
WGS analyses of the 13 isolates revealed 129 SNPs subdividing the outbreak isolates into two genome clusters, 11 ancient Beijing family isolates clustered to form major cluster, whereas two modern Beijing family isolates (No.2 and No.8) formed smaller cluster, the WGS results indicated two clonal outbreaks (Figure 1). In major cluster, the maximum number of SNPs between any pair of isolates was five SNPs, and the mean pair-wise distance (±SD) between 11 isolates was 2.57 SNPs±0.03SNPs; while isolates of the smaller cluster differed by only one SNPs in a pairwise comparison.
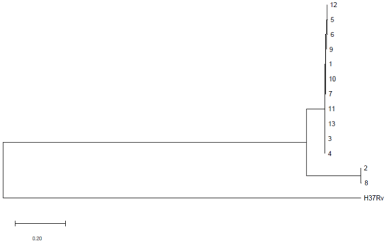
Figure 1: Maximum Likelihood tree for single nucleotide polymorphism
differences of 13 isolates isolated from high school outbreak, China.
Discussion
This study reported TB outbreak in a high school, the results highlight the importance of strengthening management of TB in school. In China, high school students suffer from stress for college entrance examination. According to Chinese guidelines for TB prevention and control in school, student with TB need to leave school for isolation and treatment, some students worry about interruption of studies rather than their disease [17]. Meanwhile, because of TB related social discrimination, some TB cases deliberately conceal their illness to avoid isolation, they may try to live with it for as long as possible, being the source of infection to others [18].The school with enclosed, air-conditioned, overcrowded classroom put students at high risk for TB infection if one student among them is infectious [3]. If the source of TB student can’t be identified immediately, this TB student will infect even more students and result in outbreak.
MIRU-VNTR typing is widely used to determine relatedness and track transmission dynamics of specific isolates of M. tuberculosis [19]. Because MIRU-VNTR analysis examines only a small proportion of M. tuberculosis genome, this method may have limited resolution [20,21]. Molecular epidemiological study in English Midlands showed MIRUVNTR typing and epidemiological risk factors are poorly predictive of close genomic relatedness when comparing to WGS, and MIRUVNTR performance varies markedly by lineage of M. tuberculosis [19]. Another study from Hawaii reported 24-loci MIRU-VNTR typing has specificity of 28.6% for identifying actual transmission clusters, and this traditional typing has poor performance especially when identifying transmission in Beijing and Manila family clusters [21]. Contrary to traditional genotyping methods, WGS Provides detailed insights into transmission dynamics [22,23]. Study in low TB incidence setting showed that WGS provides better correlation with epidemiologic data than MIRU-VNTR typing [23]. Previous studies have indicated that MIRU-VNTR typing may overestimate recent TB transmission, particularly in regions with high TB incidence settings [24, 25]. In the current study, two clonal outbreaks were supported by the results of MIRU-VNTR typing as well as WGS, but WGS provided better resolution and more detailed information for transmission of M. tuberculosis, we did not find MIRU-VNTR typing overestimated recent TB transmission, probably because small numbers of isolates were analyzed in this study.
WGS has been developed that identify transmission of M. tuberculosis by comparing genomes SNPs differences between the isolates, isolates with smaller number of SNPs differences indicate a transmission event. Currently there is no unified standard for the SNPs distance cut-off to judge possible TB transmission, and different SNPs cut off have been applied in studies in different regions. Walker et al. [26] based on studies in low TB incidence settings of the UK and propose 12-SNPs distance as the upper threshold to identify plausible transmissions and state that isolates separated by five or fewer SNPs is likely a result of recent transmission, A cut off of five SNPs or fewer for linked TB transmission is widely used in many studies [20,23,27,28]. Stucki D et al. analyzed isolates from 68 patients associated with outbreak and reported that isolates from patients with confirmed epidemiological links differ by 0-11 SNPs [29]. In our study, the maximum number of SNPs between any pair of isolates was five in clusters, which is compatible with Walker’s reports and slightly fewer than that report in other similar studies [23,30].
Study of molecular evolution in M. tuberculosis suggests strains will accumulate mutations while transmitting between patients, so the pattern of accumulated mutations can infer direction of transmission [31]. Previous findings suggested that SNPs can be used to identify transmission chains in TB clusters [26,27,32]. Contrary to previous findings, recent study has showed that WGS is able to reveal the direction of transmission of TB in small proportion of cases within the outbreak, WGS is insufficient for inferring transmission in the majority of cases, and transmission chain or network may not be useful in institutional settings, because multiple transmission events can occur with no detectable SNPs acquisition [30]. In this TB outbreak, a large number of students developed TB during a short period of time, and contact tracing investigation could not provide accurate information. WGS with high resolution was performed to delineate the chain of transmission, however, because of only 13 positive isolates were available for analysis, and the direction of transmission could not be intact established. Our data support that WGS can verify recent transmission in TB outbreak, however, it can infer transmission chain linkage only to a limited extent in TB outbreak.
One limitation of this study is that SNPs in repetitive PE/PPE/ PGRS family genes, amounting to almost 10% of the genome, was not assessed due to the difficulty in reliably calling SNPs within these regions, so it is possible we leave out mutations in these genes [33]; another limitation is the source of this outbreak remained unknown, because most students in class A senior three rent houses near the school, the spread of M. tuberculosis may be linked to high TB prevalence in the community; and the third limitation is only a few cases who had positive isolates were performed WGS analysis, so the intact transmission chains cannot be established.
Conclusion
Our findings suggest that timely identification and isolation of TB cases, especially in the early phase of outbreak, is the key to preventing spread of TB among students, and implementation of guidelines for TB prevention and control in school must be prioritized. This study demonstrates that WGS provides better resolution than MIRUVNTR genotyping for investigation of TB outbreak.
Acknowledgement
This study was supported by the national science and technology major project (No. 2018ZX10103001).
Compliance with Ethnics Guidelines
Shengfen Wang, Yi Tang, Liang Zhong, Yang Zhou, Bing Zhao, Tao Li, Qian Cheng, Yanlin Zhao and Qiaozhi Wang declare that they have no conflict of interests. This study was a response to public emergency event, ethical approval was not required, and however, all procedures in this study complied with the ethical standards of the ethical review board of the Chinese Center for Disease Control and Prevention.
References
- World Health Organization. Global Tuberculosis Report. Geneva, Switzerland: WHO. 2020.
- Ma MJ, Yang Y, Wang HB, Zhu YF, Fang LQ, An XP, et al. Transmissibility of tuberculosis among school contacts: an outbreak investigation in a boarding middle school, China. Infect Genet Evol. 2015; 32: 148-155.
- Pan D, Lan R, Graviss EA, Lin D, Liang D, McNeil E, et al. Adolescent tuberculosis associated with tuberculosis exposure in classrooms and dorm rooms in Guangxi, China. Int J Infect Dis. 2019; 78: 8-14.
- Wu X, Pang Y, Song Y, Dong W, Zhang T, Wen S, et al. Implications of a school outbreak of multidrug-resistant tuberculosis in Northern China. Epidemiol Infect. 2018; 146: 584-588.
- Fang Y, Zhang L, Tu C, Ye D, Fontaine R, Ma H, et al. Outbreak of pulmonary tuberculosis in a Chinese high school, 2009-2010. J Epidemiol. 2013; 23: 307-3012.
- Chen W, Xia Y, Li X, Zhou L, Li C, Wan K, et al. A tuberculosis outbreak among senior high school students in China in 2011. J Int Med Res 2012; 40: 1830-1839.
- You NN, Zhu LM, Li GL, Martinez L, Lu W, Liu Q, et al. A tuberculosis school outbreak in China, 2018: reaching an often overlooked adolescent population. Epidemiol Infect. 2019; 147: e303.
- National Health Commission of the People’nfectious [3]. If the source of TB sts Republic of China. Annual Report on Surveillance of Infectious Disease in China. 2017.
- Reed MB, Pichler VK, McIntosh F, Mattia A, Fallow A, Masala Set al. Major Mycobacterium tuberculosis lineages associate with patient country of origin. J Clin Microbiol. 2009; 47: 1119–1128.
- Nakanishi N, Wada T, Arikawa K, Millet J, Rastogi N, Iwamoto T. Evolutionary robust SNPs reveal the misclassification of Mycobacterium tuberculosis Beijing family strains into sublineages. Infect Genet Evol. 2013; 16: 174-177.
- Supply P, Allix C, Lesjean S, Cardoso-Oelemann M, Rüsch-Gerdes S, Willery E, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol. 2006; 44: 4498-4510.
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010; 26: 589-595.
- Chiang C, Layer RM, Faust GG, Lindberg MR, Rose DB, Garrison EP, et al. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods. 2015; 12: 966-968.
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012; 6: 80–92.
- Farhat MR, Shapiro BJ, Kieser KJ, Sultana R, Jacobson KR, Victor TC, et al. Genomic analysis identifies targets of convergent positive selection in drugresistant Mycobacterium tuberculosis. Nat Genet. 2013; 45: 1183-1189.
- Kumar S, Stecher G, Li M, Knyaz C Tamura K. MEGA X. Molecular Evolutionary Genetics Analysis across computing platforms. Molecular Biology and Evolution. 2018; 35: 1547-1549.
- Zhang S, Li X, Zhang T, Fan Y, Li Y. The experiences of high school students with pulmonary tuberculosis in China: a qualitative study. BMC Infect Dis. 2016; 16: 758.
- Abebe G, Deribew A, Apers L, Woldemichael K, Shiffa J, Tesfaye M, et al. Knowledge, health seeking behavior and perceived stigma towards tuberculosis among tuberculosis suspects in a rural community in southwest Ethiopia. PLoS One. 2010; 5: e13339.
- Wyllie DH, Davidson JA, Grace Smith E, Rathod P, Crook DW, Peto TEA, et al. A Quantitative Evaluation of MIRU-VNTR Typing Against Whole-Genome Sequencing for Identifying Mycobacterium tuberculosis Transmission: A Prospective Observational Cohort Study. EBioMedicine. 2018; 34: 122-130.
- Roetzer A, Diel R, Kohl TA, Rückert C, Nübel U, Blom J, et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med. 2013; 10: e1001387.
- Koster KJ, Largen A, Foster JT, Drees KP, Qian L, Desmond E, et al. Genomic sequencing is required for identification of tuberculosis transmission in Hawaii. BMC Infect Dis. 2018; 18: 608.
- Gardy JL, Johnston JC, Ho Sui SJ, Cook VJ, Shah L, Brodkin E, et al. Wholegenome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011; 364: 730–739.
- Alaridah N, Hallbäck ET, Tångrot J, Winqvist N, Sturegård E, Florén- Johansson Ket al. Transmission dynamics study of tuberculosis isolates with whole genome sequencing in southern Sweden. Sci Rep. 2019; 9: 4931.
- Stucki D, Ballif M, Egger M, Furrer H, Altpeter E, Battegay M, et al. Standard Genotyping Overestimates Transmission of Mycobacterium tuberculosis among Immigrants in a Low-Incidence Country. J Clin Microbiol. 2016; 54: 1862-1870.
- Sloot R, Borgdorff MW, de Beer JL, van Ingen J, Supply P, van Soolingen D. Clustering of tuberculosis cases based on variable-number tandem-repeat typing in relation to the population structure of Mycobacterium tuberculosis in the Netherlands. J Clin Microbiol. 2013; 51: 2427-2431.
- Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, et al. Wholegenome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013; 13: 137-146.
- Kato-Maeda M, Ho C, Passarelli B, Banaei N, Grinsdale J, Flores L, et al. Use of whole genome sequencing to determine the microevolution of Mycobacterium tuberculosis during an outbreak. PLoS One. 2013; 8: e58235.
- Meehan CJ, Moris P, Kohl TA, Pecerska J, Akter S, Merker M, et al. The relationship between transmission time and clustering methods in Mycobacterium tuberculosis epidemiology. EBioMedicine. 2018; 37: 410-416.
- Stucki D, Ballif M, Bodmer T, Coscolla M, Maurer AM, Droz S, et al. Tracking a tuberculosis outbreak over 21 years: strain-specific single-nucleotide polymorphism typing combined with targeted whole-genome sequencing. J Infect Dis. 2015; 211: 1306-1316.
- Casali N, Broda A, Harris SR, Parkhill J, Brown T, Drobniewski F. Whole Genome Sequence Analysis of a Large Isoniazid-Resistant Tuberculosis Outbreak in London: A Retrospective Observational Study. PLoS Med. 2016; 13: e1002137.
- Schürch AC, Kremer K, Kiers A, Daviena O, Boeree MJ, Siezen RJ, et al. The tempo and mode of molecular evolution of Mycobacterium tuberculosis at patient-to-patient scale. Infect Genet Evol. 2010; 10: 108-114.
- Schürch AC, Kremer K, Daviena O, Kiers A, Boeree MJ, Siezen RJ, et al. High-resolution typing by integration of genome sequencing data in a large tuberculosis cluster. J Clin Microbiol. 2010; 48: 3403-3406.
- McEvoy CR, Cloete R, Müller B, Schürch AC, van Helden PD, Gagneux S, et al. Comparative analysis of Mycobacterium tuberculosis pe and ppe genes reveals high sequence variation and an apparent absence of selective constraints. PLoS One. 2012; 7: e30593.