
Research Article
J Bacteriol Mycol. 2019; 6(6): 1119.
Inhibition of Macrophage Activity and Expression Profile of IL Genes in Goldfish after Challenge and Immunized with Aeromonas hydrophila
Devia G1, Balasundaramb CH2 and Ramasamyc H3*
¹Department of Zoology, Nehru Memorial College, Puthanampatti 621 007, Tamil Nadu, India
²Department of Herbal and Environmental Science, Tamil University, Thanjavur 613 005, Tamil Nadu, India
³Department of Zoology, Pachaiyappa’s College for Men, Kanchipuram - 631 501, Tamil Nadu, India
*Corresponding author: Ramasamyc H, Department of Zoology, Pachaiyappa’s College for Men, Kanchipuram - 631 501, Tamil Nadu, India
Received: November 27, 2019; Accepted: December 26, 2019; Published: December 31, 2019
Abstract
We investigate and characterized the macrophage activating and deactivating cytokines in mammalian systems about these immunoregulatory molecules in fish. We partially purified Macrophage Deactivating Factor (MDF) from mitogeninduced goldfish kidney leukocytes using gel permeation and chromatofocusing fast performance liquid chromatography (GP-FPLC and C-FPLC). The pretreated macrophages for 6 or 24 h with MDF before activation with Macrophage Activating Factors (MAF) and/or bacterial Lipopolysaccharide (LPS) exhibited a down-regulation in their NO response. However, treated with MDF on 24 h did not activation with MAF and LPS. The MDF treatment is impaired the NO response of goldfish macrophages infected with the mammalian protozoan parasite Leishmania major. Therefore, the present results suggest that MDF exhibits its inhibitory effect downstream of the converging intracellular pathways induced by LPS and/or L. major. In addition to investigate differential constitutive expression of IL-1β1, IL-1β2 and IL-6 genes in kidney, intestine, and spleen of goldfish (Carassius auratus) after challenge and immunization with Aeromonas hydrophila using real-time PCR analysis. All the tested interleukin gene mRNA expression levels higher in kidney, intestine, and spleen fish were injected with heat-killed or formalin-killed vaccines. However, most of the tissues a modest down-regulation in expressions of infected untreated fish. Therefore, our results indicate that vaccines treated fish up-regulation in expressions in tissues could be central regulatory and effector cytokine of inflammatory and antimicrobial responses.
Keywords: Aeromonas hydrophila; Carassius auratus; Macrophage activity; Interleukin genes; Vaccines
Introduction
In fish, many interleukins e.g. IL-1 [1], IL-2 [2], IL-6 [3], IL-8 [4], IL-10 [5], IL-11 [6], type 1 and type 2 interferons [7], lymphotoxin β [8] have been identified and cloned. Recently, IL-1β [9] have been cloned and sequenced in rainbow trout and recombinant proteins produced to study their respective functions [10]. IL-1β is a proinflammatory cytokine gene that directly stimulating the innate immune system during later stages of infection [11]. The important role of IL-1β is activation of T and B cells [12]. The IL-1β produced as a precursor molecule is cleaved to generate a mature peptide that affects most cells and immune organ systems.
The activation of specific Pathogen Recognition Receptors (PRRs), molecular moieties pathogen-specific immune responses are coordinated and dependent present upon sub-sets of leukocytes, such as macrophages or dendritic cells. The PRRs are respond to pathogens or their Pathogen Associated Molecular Patterns (PAMPs) by the initiation of distinct transcriptomic programmes, which will dictate the cellular or tissue response [13,14]. In mammals, the host transcriptional programmes have been identified by microarray analysis for specific PAMPs to bacterial [15], viral [16], parasitic [17], and fungal infections [18]. Both macrophages and dendritic cells are initiates the immune response by secreting molecules, such as proinflammatory cytokines [19]. These arrays have been used to study the response in fish to vaccination [20] or stimulation with LPS [21].
The macrophage response to infection or activation by immune stimulants can be effectively analysed by microarray that allowing thousands of genes to be monitored for expression in parallel [22]. These microarrays employed by pathogens to evade the immune system are complex and by studying specific cell types or tissues the host defence strategies. In addition the gene expression response of T cells to PAMPs has been explored using microarrays [23]. To further characterize the response of specific cytokines have been used to stimulate human [24], murine [25], and bovine [26] macrophage cell lines. The availability of salmonid-specific gene chips [27- 29] has provided the means to begin to characterise the salmonid immune response at a global gene level both in vitro and in vivo. This technology will afford a deeper understanding of overall cellular and tissue processes during immune activation. A number of recent reports concerning PAMPs recognition [30], activated macrophage transcriptomics [29], immunomics [30,31], and genome-wide surveys [32,33] showed that fish and fish macrophages should lead to different physiological/immunological responses due to pathogens in vivo.
The head kidney can consequently integrate the neuro-immunoendocrine milieu in normal and pathological states. However, few global gene regulation studies concerning the molecular regulation of head kidney function during infection or PAMPs stimulation in salmonids [34] have been described. Although many studies have used this tissue as a primary source of macrophage-like cells the activation of the immune systems [35]. Lipopolysaccharides (LPS) is the major constituent of Gram-negative bacteria is widely used for PAMP-preparation, which induces potent immune responses. A portion of LPS molecule is primarily responsible for the endotoxic properties in animals [36,37].
Intracellular killing of microorganisms by macrophages is essential for protection against a variety of pathogens including protozoan parasites, fungi, bacteria, and viruses [38-42]. Cytokine-activated vertebrate macrophages kill these pathogens by producing a number of highly toxic molecules including Reactive Oxygen Intermediates (ROI) and Reactive Nitrogen Intermediates (RNI) [43-45]. Nitric oxide (NO ) is reactive nitrogen intermediate produced in the cytoplasm of macrophages through the enzyme catalyzed oxidation of the terminal guanido nitrogen of L-arginine [45] and freely diffuses across cell membranes to target enzymes that contain catalytically active labile iron [40]. Production of NO by activated macrophages appears to be a primitive killing mechanism since immunocytes from invertebrates such as insects and starfish have also been reported to produce NO [46,47] that activated Goldfish Macrophage Cell Line (GMCL) [48] and primary goldfish macrophages [49].
Aeromonas hydrophila is well known to cause a variety of diseases in fish including goldfish, such as haemorrhagic septicaemia, infectious dropsy, tropical ulcerative disease and fin rot leading to heavy mortality in aquaculture industry [50,51]. Various synthetic chemicals and antibiotics have been used to prevent or treat fish diseases with a partial success. Treatment with adjuvanted vaccine is one such strategy as the successful development of new vaccines. It is reliant upon the availability of adjuvants that are not only safe for the host, but also induce immune responses complementary during natural infection [52]. Immunostimulants, when used alone to increase the immunocompetence and disease resistance of fish by enhancing the nonspecific defence mechanisms [53]. The adjuvants used in vaccines preparations that activate antigen-presenting cells (e.g. macrophages) to produce more of the signal molecules (e.g. cytokines) to recruit other immune system cells [54]. However, very few reports, but there is no report in goldfish against A. hydrophila infection.
The aim of this paper to investigation is to characterize in vivo biological activities displayed by goldfish IL-1β1, IL-1β2 and IL-6 genes using an immunologically tractable model to focus special attention is mainly involved in the recruitment of leukocytes to the inflammatory foci rather than in their activation. In addition to investigate the release of cytokines from head kidney (HK) leucocytes susceptibility in fish after challenge and immunization (heatkilled and formalin-killed) with A. hydrophila in goldfish and their inhibition of macrophage activity and differential tissue expression by RT-PCR.
Materials and Methods
Fish
Healthy goldfish, Carassius auratus weighing approximately 38 g were purchased from a local fish farm in Jeju Island, South Korea and transported to the laboratory in plastic bags filled with oxygenated water. The fishes were maintained randomly into 150-L aquaria a total of 400 fish. All the fish were acclimated for 2 week under laboratory conditions (14/10 h light/dark cycle) prior to challenge or immunization. The aquaria water quality parameters were monitored during the experimental period as dissolved oxygen concentration 5.5 - 7.4 mg l-1 (Winkler’s method), pH 5.6 - 7.3, and temperature at 18 - 21°C. Fish were fed with a standard pelleted diet at 3% of their body weight twice a day during the experiment. Water of the aquarium was exchanged partially daily to remove waste feed and faecal materials.
A. hydrophila
A. hydrophila (KCTC 2358) was obtained from Korean Collection for Type Cultures (KCTC) in Daejeon, South Korea and maintained in the laboratory. Subcultures were maintained on tryptic soy agar (TSA, Sigma) in slopes at 5°C and routinely tested for pathogenesis [55], by inoculation into goldfish [56]. Stock culture in tryptic soy broth (TSB, Sigma) was stored at -70°C in 0.85% NaCl with 20% glycerol (v/v) to provide stable inoculate throughout the experiment [57]. Subculture of A. hydrophila was taken on TSA slope and harvested by TSB. The inoculated TSB was incubated for 24 h in a shaker at 30°C, and then centrifuged at 12000 g for 10 min at 4°C [57]. The supernatant was discarded and the bacterial pellet was washed three times with Phosphate-Buffered Saline (PBS) at pH 7.2. The number of A. hydrophila cells ml-1 in one day culture was enumerated using standard plate count methods on TSA plates supplemented with 5% sheep’s blood [57]. An aliquot of 25 μl of culture used in the challenge was plated on BHI agar plates and incubated for 48 h at 28°C.
Preparation of vaccines
The whole-cell bacterin was prepared by Akhlaghi et al [58] with some modifications. A. hydrophila were grown for 48 h at 28°C in TSB, and then washed with PBS for three times. Bacteria were grown to a density of approximately 1.0 x 105 viable cells ml-1. Suspensions containing bacterial cells were treated with formalin to a final concentration of 0.4% (v/v) overnight at 4°C. The suspension was centrifuged and washed three times with PBS as the initial volume, checked the sterility of bacteria and then stored at -70°C until use. The washed, Formalin-Killed bacterial Cells (FKC) were resuspended in PBS and stored at 4°C until used. Heat-Killed bacterial Cells (HKC) were obtained by subjecting the harvested cells to 100°C for 15 min. Both FKC and HKC thereafter diluted with an equal volume of Freund’s complete adjuvant (FCA; ICN Biomedicals). The vaccines were stored at 4°C until use. Before use, the vaccines were kept at room temperature. The efficiency of E-mediated killing of A. hydrophila bacteria was estimated by plating samples of appropriate dilutions of freshly harvested FKC and HKC agar [59], and results were compared with those from samples obtained prior to onset of lysis. Results indicated a 100% killing efficiency as no colony forming units (cfu) were found on plates.
Experimental design, immunization and cumulative mortality
Four groups of goldfish (n = 400), each comprising 50 fish in triplicate. Before injection, all fish were anaesthetised in tricaine methanesulfonate (MS222, Sigma) (100 mg l-1). One of the group Formalin-Killed Vaccine (FKV) was immunized against A. hydrophila by intra-pritoneal injection with 0.2 ml of Formalin-Killed Bacteria (FKB). Another group Heat-Killed Vaccine (HKV) was immunized against by intra-pritoneal injection with 0.2 ml of Heat-Killed Bacteria (HKB). After 15 days fish received same volume of FKB or HKB as a booster dose. Infected untreated group (I) were injected with 0.1 ml PBS containing A. hydrophila at a concentration of 1.0 x 105 viable cells ml-1. The Control group (C) was injected with 0.2 ml sterile PBS or FCA. Earlier the challenge dose was standardized to give 90% mortality in the infected untreated group (I). The cumulative mortality of control or experimental (each in 20 fish) were recorded daily basis for 30 days. Relative Percent Survival (RPS) was calculated by the following formula of Amend [60],
RPS (%) = 1 – ((% test mortality) x 100)) / (% control mortality)
Sample collection
The anterior kidney, spleen, and intestine tissue sample were collected in triplicate aquaria per group per treatment (control or experimental) on 30 days. Fish were anesthetized in a 100 mg l-1 solution of tricaine methanesulfonate (MS-222, Syndel) before collection of kidney leucocytes and tissue samples. Individual fish was sampled only once to avoid the influence on the assays due to multiple bleeding and handling stress on the fish. All tissue samples were rinsed in cold phosphate buffered saline (PBS, Gibco) at pH 7.2 and stored in 1-ml Trizol® (Invitrogen) frozen at -80°C in liquid nitrogen until DNA or RNA extraction. The Head Kidney (HK) leucocyte cells were subsequently removed for bioassay as described below.
Growth medium and isolation of goldfish kidney leukocytes
The complete culture growth medium contained 5% carp serum and 10% fetal calf serum (Hyclone) used in all experiments has been previously described by Neumann et al. [44]. Head kidney leukocytes were isolated from goldfish kidneys following Neumann et al. [49]. Kidneys were aseptically removed and placed into a petri dish containing ice-cold medium. Using a sterile plunger from a 3cc syringe, kidneys were gently pressed through sterile stainless steel screens to release kidney cells. Screens were rinsed with medium containing antibiotics, such as 50 μg ml-1 of gentamicin, 100 U ml-1 of penicillin, 100 μg ml-1 of streptomycin, and 50 U ml-1 of heparin. The resulting cell suspension was layered on 51% Percoll (Pharmacia) and centrifuged at 400 g for 25 min. Cells at the medium-51% Percoll interface were removed with a sterile pipette and transferred to sterile centrifuge tubes. To remove Percoll, cells were washed twice in serum-free medium and again centrifuged at 200 g for 10 min at 4°C. The viable leukocytes were enumerated using a haemocytometer after staining with trypan blue (Gibco).
Generation of in vitro-derived kidney macrophages (IVDKM)
The goldfish kidney leukocytes and macrophages secrete growth factors that induce selective proliferation and differentiation of macrophages from kidney hematopoietic tissues of the goldfish [61,62]. Cell Conditioned Medium (CCM) containing macrophage growth factors were obtained from the supernatants of 8-10 day old kidney leukocyte cultures. Kidney leukocytes (15-20 x 106 cells) were cultured in 20 ml of complete medium supplemented with 25% CCM. Cells were incubated at 20°C and fed on day 5 with 5 ml of complete medium. Cultures, 8-10 days old, were used as a source of macrophages for bioassays. Supernatants from these cultures were used as a source of CCM for establishing new macrophage cultures.
Generation of leukocyte supernatants for cytokine activity
Crude cytokine preparations were established following Neumann et al. [61]. The kidney leukocytes isolated from 25 fish were pooled and seeded in 75 cm2 tissue culture flasks at a concentration of 4 x 106 cells/ml, and incubated overnight in medium containing 2.5% carp serum and 10% fetal calf serum (Hyclone) at 20°C. Mixed leukocyte cultures were stimulated the following morning with 10 μg/ml concanavalin A (Con A, Boehringer Mannheim), 10 ng ml-1 phorbol myristate acetate (PMA, Sigma), and 100 ng ml-1 calcium ionophore A23187 (Sigma). Cultures were stimulated with these mitogens for 6h, after which the mitogens and serum were removed by washing the adherent cell layer with three changes of 20 ml Hanks Balanced Salt Solution (HBSS). The remaining adherent cell layer was given fresh serum-free medium and incubated for 72h at 20°C. Supernatants were subsequently removed, filter sterilized, and stored at -20°C until used in assays. These cytokine preparations were used as either a source of Macrophage Activating Factors (MAFs), or for isolating goldfish Macrophage Deactivating Factors (MDFs).
Characterization of MDF
Gel-permeation fast performance liquid chromatography (GPFPLC): The initial analyses of MAF and MDF activities were following Neumann et al. [61]. The cytokine preparations were concentrated by dialysis against Polyethyleneglycol (PEG) and were placed into dialysis bags (molecular weight cutoff=3.5 kD, SpectroPor) and covered in PEG flakes (MW=20 kD, Sigma). Concentration was allowed to proceed until half of the original volume remained in the dialysis bag. Samples underwent repeated half concentrate dialysis until the volume of the original crude preparation was concentrated 36-fold. The concentrated cytokine preparations were filter sterilized (0.22 mM filter, Millipore), separated into 500 ml samples, and stored at -20°C. Then the cytokine samples were fractionated according to size using a Superose 6 column (Pharmacia). GP-FPLC was carried out at 22°C using an FPLC system from LKB (Pharmacia, Bromma, Sweden). Concentrated cytokine preparations were thawed and centrifuged at 19000 g for 10 min before injecting 200 ml fractions onto the column. The running buffer used for GP-FPLC was 1x PBS (pH 7.2). All GP-FPLC fractions were collected at 2.5 min intervals into 15 ml centrifuge tubes, subsequently sterilized, and stored at -20°C until used in assays.
Chromatofocusing gel- permeation fast performance liquid chromatography (C-FPLC): Separation of GP-FPLC fractions by isoelectric focusing was performed following Neumann et al. [61]. GPFPLC fractions displaying maximal MDF activity were concentrated using microcentrifugal concentrators (Filtron; MW cutoff=3 kD). Prior to addition of MDF, the polystyrene microcentrifugal sample chamber was blocked for 30 min with 1% calf serum to prevent non-specific absorption of MDF activity. Chromatofocusing of concentrated MDF was performed using a Mono-P column (Pharmacia). The Mono-P column was pre-equilibrated with 0.025 M bis-Tris (pH 7.0, 1 M HCL) for establishment of the upper limits of the gradient. A linear descending pH gradient (7.0-4.0) was established by running a 1:10 dilution of Polybuffer 74 (Pharmacia) at a flow rate of 0.75 ml/min through the column. MDF samples (500 ml) were allowed to elute through the column for 30 min prior to initiation of the pH gradient. C-FPLC fractions were collected in 15 ml polystyrene tubes containing an equal volume of 10% calf serum (diluted in 1x PBS) in order to stabilize biological activity. Proteins bound to the Mono-P column (i.e. proteins having an isoelectric point of less than 4.0) were eluted from the column using a 2 M NaCl solution. This salt solution was injected onto the Mono-P column (5 injections of 500 ml) at flow rate of 0.25 ml/min. Protein elution was monitored by UV absorption (280 nm), and approximately 2 ml of eluted protein was collected. Of the eluent, 1 ml was stabilized by adding an equal volume of 10% calf serum (in PBS), and placed in a dialysis bag (3.5 kD cutoff, SpectroPor). This sample was dialyzed overnight in 1x PBS to remove excess salt. The serum-stabilized SE was subsequently tested for MDF activity using the NO bioassay.
Functional characterization of MDF in crude cytokine preparations
The functional analysis of GP-FPLC crude cytokine preparations was determined by pre-treating 8-10 days old IVDKM (5 x 104 cells/ well) with 25 ml of each fraction (1:3 dilution) for 6 h. Cells were subsequently activated with LPS (1 μg ml-1) and crude MAF (1:4 dilution) and NO production determined 72 h later using the Griess reaction.
MDF inhibition of activated goldfish macrophage NO responses
To assess the ability of MDF to inhibit NO production of activated macrophages, 8-10 day old IVDKM were seeded into the wells of half-area 96-well culture plates (Costar) at 5 x 104 cells/well and pretreated for 6 h with GP-FPLC fraction containing maximal MDF activity (1:3 dilution), or C-FPLC SE (1:5 to 1:160). Macrophages were subsequently activated with crude MAF (1:4) and LPS (1 μg ml- 1), LPS alone (1 and 10 μg ml-1), or infected with Leishmania major. Activated macrophages were then incubated for an additional 72h at 20°C before determination of nitrite production by the Griess reaction.
Effects of activation sequence and MDF dose on NO production
On 8-10 days old IVDKM were exposed to MDF at varying times pre- and post-activation to determine whether macrophages required pre-treatment with MDF in order to deactivate nitric oxide production by activated macrophages. Goldfish macrophages were placed in wells of half-area 96-well culture plates (5 x 104 cell/ well), and triplicate groups treated with the GP-FPLC fraction with maximal MDF activity (1:5 dilution) 24 or 6h prior to activation with MAF (1:4) and LPS (1 μg ml-1). In parallel cultures, macrophages were treated with MAF (1:4) and LPS (1 μg ml-1) for 24 h prior to addition of MDF (1:3 dilution). Then the macrophages were incubated for 72h after stimulation at 20°C before determination of nitrite production by Griess reaction. The effect of MDF dose on inhibition of NO production by goldfish macrophages was determined by plating 5 x 104 goldfish macrophages in wells of half-area 96-well culture plates and pre-treating macrophages for 6h with serial dilutions of MDF (from 1:5 to 1:160). The macrophages were subsequently activated with MAF (1:5) and LPS (1 μg ml-1) or LPS alone (1 and 10 μg ml-1) and incubated for 72h at 20°C before determination of NO production.
Nitric oxide assay
NO production by goldfish macrophages was determined indirectly using the Griess reaction [62]. A volume of 75 μl of supernatants from individual macrophage cultures was transferred to a microtitre plate and 100 μl of 1% sulfanilamide (Sigma) (dissolved in 2.5% H3PO4) followed by 100 μl of 0.1% N-naphthyl-ethylenediamine (Sigma) (dissolved in 2.5% phosphoric acid) was added to each well. The plate was allowed to sit for 2 min before the optical densities (OD 540 nm) were determined using an automated microtitre plate spectrophotometer (Biotek). The approximate concentration of nitrite in samples was determined from a standard curve generated using known concentrations of sodium nitrite.
Effects of MDF treatment on the viability
Goldfish macrophages were seeded in triplicate into 6 ml tubes at a cell density of 2.5 x 105 cells in 250 μl. Cells were pre-treated with 125 μl medium, 1x PBS at pH 7.2, or partially purified MDF for 6h. Subsequently the macrophages were activated with MAF (1:5 final dilution) and LPS (1 μg ml-1) and incubation at 20°C for 72h. The nitrite production was determined in the culture supernatants and cell number and viability by haemocytometer after staining with trypan blue (Gibco).
Isolation of total RNA and cDNA synthesis
Tissue samples were subsequently thawed and homogenized in RNAzol B (Biogenesis) on ice. Total RNA was extracted and reversed transcribed analysis were taken an equal amount (50 mg) of tissue samples was obtained separately from each tissue in three replicate to make a pool, before isolation of the RNA. Total RNA was extracted from pooled tissue (150 μg) using Trizol® (Invitrogen) in according to the manufacturer’s protocol. The total RNA was stored at -80°C until further use. The total RNA concentration and purity were determined by measuring the absorbance at 260 and 280 nm in a UV-spectrometer (Bio Rad, USA). Originally purified RNA was diluted up to 1 μg/μl concentration before synthesis of cDNA. Two micrograms of total RNA of goldfish tissues were used to synthesize cDNA from each tissue using a Superscript™ III first-strand synthesis system for RT-PCR kit (Solgent). Then the RNA was incubated with 1 μl of 50 mM oligo (dT) 20 (500 μg ml-1, Invitrogen) and 2 μl of 10 mM dNTPs (Solgent) for 10 min at 70°C. After incubation, 4 μl of 5x cDNA synthesis buffer (Solgent), 1 μl of dithiothreitol (DTT, 0.1 M, Solgent), 0.5 μl of RNase inhibitor (Solgent) (40 U/μl), and 1 μl of SuperScript™ III reverse transcriptase (15 U/μl) were added and incubated for 1h at 50°C. Then, 1 μl Diastar RNase was added to each cDNA and incubated at 50°C for 50 min. Finally, synthesized cDNA was diluted 10-fold (total 200 μl) before storing at -20°C.
mRNA expression analysis by real time PCR
PCR was carried out using different primers sets and different conditions for β-actin (positive control), IL-1β1, IL-1β2, and IL-6 genes were given in Table 1. The β-actin and IL-1β1, IL-1β2, and IL-6 PCR’s, amplifications were performed in 25 μl reactions containing the following components: 5 ml of cDNA template (diluted in water), 1 μl (25 pmol) of each primer, 2.5 ml of 10X reaction buffer (160 mM (NH4)2SO4, 670 mM Tris–HCl, pH8.8, 0.1% Tween-20, Bioline), 0.5 ml dNTP mixture (2.5 mM for each base, Bioline), 1.25 μl of 50mM MgCl2 (Bioline), 0.125 μl (0.625 U) of Taq polymerase (Bioline) and 13.625 ml of sterile H2O The components of the PCR reaction for IL 1β1, IL-1β2, and IL-6 were identical, except that 1.25 μl forward and reverse primers, 1 μl dNTP mix, 1 μl MgCl2 and 13.375 μl of H2O was used. The first PCR in each case was for β-actin, and the amount of cDNA used in each sample was titrated (between 1 and 3 μl) to give a constant product yield. The same amount of cDNA was then used for all subsequent immune gene PCR’s as a way of normalising the data in order to give a more quantitative result.
![]()
Genes
GenBank accession no.
Primer sequence
Size (bp)
Tem. (°C)
Cycles
Time
β-actin
BAA92339
F: ATTGTGATGGACTCCGGTGATGGT
386
95
1x
2 min
R: AAGGTGGTCTCATGGATACCGCAA
95
30x
20 s
64
30x
40 s
72
30x
1 min
72
1x
5 min
IL-1β1
AJ419848
F: TCCTCACAGCATGAAGAAGGTGGT
419
95
1x
2 min
R: ACCCATCAGACTCGGTACAAGCAA
95
30x
20 s
64
30x
40 s
72
30x
1 min
72
1x
5 min
IL-1β2
AJ419849
F: ATAAGACCAGGCAGACCTTGCAGT
450
95
1x
2 min
R: TTGGCCTCTGGTACATTTCCACCT
95
30x
20 s
64
30x
40 s
72
30x
1 min
72
1x
5 min
IL-6
DQ861993
F: AGGCTCACCAGGTTAACGAGCAAA
352
95
1x
2 min
R: TTTCAGCTGGCTCAGGAATGGGTA
95
30x
20 s
64
30x
40 s
72
30x
1 min
72
1x
5 min
Table 1: Gene-specific primers sets and conditions used for PCR.
Analysis of expression profile and the relative expression ratio
PCR products were visualised on a 2% agarose gel containing 0.1 mg ml-1 ethidium bromide. The relative levels of RNA were quantified for each gene by densitometric scanning of agarose gel images using a UVP gel imaging system and UVP Gel-works ID advanced software. Ratios of gene product: β-actin product were calculated for each of the three genes (for IL-1β1, IL-1β2, and IL-6) and used to quantify inter-group differences in expression levels. The products were run on the gel for 1h at 100 V, using a 100 bp DNA ladder (Bioneer) as a size marker. All the gel was then visualized using a Gel Doc image analysis system (Bio-Rad). The relative folds (ratio) of IL-1β1, IL- 1β2, and IL-6 expression relative to β-actin expression using the pixel density for each product were determined by Lab Works image acquisition and analysis software. This enabled the evaluation of differential expression of IL-1β1, IL-1β2, and IL-6 genes between different sample groups.
Statistical analysis
The PBS injected sample as expression was compared challenged or immunized with A. hydrophila samples to determine the relative level of expression and respective PBS control for each induction experiment to determine the fold change in expression. All data represent means ± standard error and were subjected to a oneway analysis of variance (ANOVA) followed by Duncan’s Multiple Range test using the SPSS 11.5 program. Differences were considered statistically significant at p ‹ 0.05.
Results
Inhibition of macrophage activity
The effect of Macrophage Deactivation Factor (MDFs) on viability cells in the medium, Control (C), Infected (I), Heat-Killed (HKV) and Formalin-Killed (FKV) vaccines treated groups are show in (Figure 1). The viability cells in the medium were 50.0±0.58. The control group the viability cells were high at 54.5±0.50. However, infected untreated group was low at 51.8±0.79. On the other hand, heat-killed and formalin-killed vaccine treated groups were 55.2±0.75 and 58.3±0.49 (Figure 1).
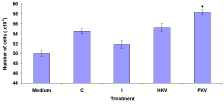
Figure 1: Effect of macrophage deactivation factor (MDFs) on viability cells in
the medium, Control (C), Infected (I), Heat-Killed (HKV), and Formalin-Killed
(FKV) vaccines treated groups.
The percentage of total cell number cells in the medium of Macrophage Deactivation Factor (MDFs) was 86.8±0.65. The percentage of total cell number cells in the control group was high at 89.8±0.65. But infected untreated group, the percentage of total cell number cells was low at 88.3±0.56. On the other hand, heat-killed and formalin-killed vaccine treated groups were high at 90.8±0.79 and 93.5±0.89 (Figure 2).
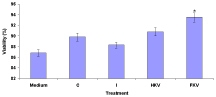
Figure 2: Effect of Macrophage Deactivation Factor (MDFs) on viability (%)
of total cell number cells in the medium, Control (C), Infected (I), Heat-Killed
(HKV), and Formalin-Killed (FKV) vaccines treated groups. % Viable cells =
number of live cells - number of dead cells / number live cells x 100.
The nitric oxide production in the medium was 1.15±0.08. The control group, the nitric oxide production was very high at 13.4±0.32. However, infected untreated group the nitric oxide production was very low at 3.5±0.13. Interestingly, heat-killed and formalin-killed vaccine treated groups were high at 8.25±0.26 and 10.4±0.38 (Figure 3).
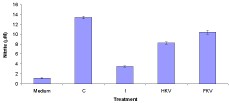
Figure 3: Nitric oxide production in the medium, Control (C), Infected (I),
Heat-Killed (HKV), and Formalin-Killed (FKV) vaccines treated groups of
stimulated goldfish Macrophages (MDFs).
GP-FPLC partially purified MDF inhibited the NO response of macrophages activated with different doses of LPS. The MDF also significantly inhibited the NO production of goldfish macrophages activated with MAF and LPS and that of goldfish macrophages infected with A. hydrophila (Figure 4). The NO response of infected or immunized or cytokine-activated macrophages was abrogated by serial dilution of the MDF, indicating a dose-dependency for MDF activity (Figures 4 and 5).
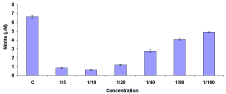
Figure 4: Nitric oxide production of goldfish macrophages (5 x 104 cells/well)
following infection with A. hydrophila were seeded into half-area 96-well
culture plates (Costar). Cells were then treated with various concentrations of
MDF (1:5±1:160 final concentration) or medium 6h prior to addition of bacteria
(mean ± SEM in triplicate cultures and is representative of two independent
experiments that were performed).
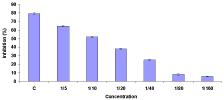
Figure 5: Macrophage (5 x 104 cells/well) Deactivation Factor (MDF)
dose response were seeded into half-area 96-well culture plates (Costar)
and pre-treated for 6 h with various MDF concentrations (1:5±1:160 final
concentration). Macrophages were subsequently activated with MAF (1:5)
and LPS (1 mg ml-1) and cells were incubated for 72 h following stimulation at
20°C prior to determination of nitrite production (% inhibition nitrite production
of triplicate cultures and is representative of two experiments that were
performed).
Expression after post-challenge and immunization with A. hydrophila in goldfish tissues
The tissue-specific IL-1β1, IL-1β2, and IL-6 genes expression were measure by semi quantitative Real-Time PCR. The IL-1β1 of control group’s expression was high in the kidney and low in the spleen tissues. The IL-1β2 expression was high in the spleen and low in the intestine tissues of control groups. On the other hand, the control group IL-6 genes expression was similar both in kidney and spleen tissues. The infected untreated group, the IL-1β1 expression was very low in the spleen and other tissues little or no expression. On the other hand, IL-1β2 expression was low in the kidney and other tissues are absent. The heat-killed vaccine treated group, the IL-1β1 expression was high in the kidney tissues while the IL-1β2 expression in the spleen. The other tissues of the IL-1β1 and IL-1β2 expression was very low or absent. However, the IL-6 genes expression was seen high all the tissues. The formalin-killed vaccine treated group of all genes were seen the expression. However, the expressions were high in the kidney and spleen then the intestine (Figure 6).
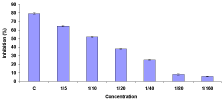
Figure 6: Real time PCR results of IL-1β1, IL-1β2, and IL-6 genes expression
in the Control (C), Infected (I), Heat-Killed (HKV), and Formalin-Killed (FKV)
vaccines treated goldfish kidney, intestine, and spleen. The expression of
each gene was normalised to β-actin. The real time PCRs were all performed
in triplicate and are shown as mean ± SEM.
Disease resistance
The Infected untreated goldfish (I) the cumulative mortality was 90% for 30 days post-challenge. However, infected goldfish after immunized with Heat-Killed or Formalin-Killed (HK or FK) A. hydrophila treated goldfish mortality were 35% and 25%. The Control (C) group without challenge with A. hydrophila group has no mortality (Figure 7).
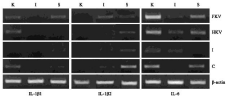
Figure 7: Cumulative mortality of control (C), Infected untreated (I), Heat-
Killed (HKV) or Formalin-Killed (FKV) A. hydrophila vaccines administration
for 30 days.
Discussion
The mitogen-induced fish kidney leukocyte supernatants to be produce macrophage activate factors that induce macrophage antimicrobial responses in goldfish. The crude cytokine preparations containing factors, such as activate and deactivate macrophage antimicrobial responses [63]. Crude cytokine supernatants contain 30 and 50 kD molecules that induce the production of ROI and RNI in fish macrophages, while 12 kD molecule inhibits RNI production [63]. The present study further understanding the biological activity of this MDF, and describes the partial purification of this molecule from crude cytokine preparations in goldfish. The MDF from crude cytokine preparations required that these crude preparations first be fractionated using GP-FPLC in order to separate MAF activity from MDF activity. The supernatants collected from mitogen-stimulated leukocytes were purified GP-FPLC and individual fractions tested for their ability to inhibit NO production of activated macrophages. The macrophages were pre-treated with purified GP-FPLC fractions prior to stimulation with MAF and LPS, ensuring that MDF would be presented to macrophages prior to activation with MAF and/or LPS.
The crude cytokine preparations are present MDF that inhibited NO production of goldfish macrophages and stimulated with MAF and LPS or LPS only. Simultaneous addition of MDF and the activating signals caused only a 34% reduction in the macrophage NO response. However, macrophages treated with MDF 24h after stimulation with MAF and LPS, exhibited normal NO production. This optimal MDF activity is achieved by pre-treating with MDF prior to activation of macrophages. These results are similar to those reported for murine macrophages treated with IL-4 shown to inhibit NO production by IFN-γ- and/or LPS activated macrophages provided that macrophages were pre-treated with IL-4 prior to activation with IFN-γ and/or LPS [62]. Interleukin 4 added 18 h after IFN-*γ and/or LPS has been shown to enhance nitric oxide production and killing of L. major by activated murine macrophages [64].
This study goldfish macrophages were mount potent NO responses when infected with A. hydrophila. It is well established that infected murine macrophages require an accessory stimulus, such as IFN-γ, for induction of their NO response [40,65]. These results suggest that fish macrophages may produce NO in response to an intracellular infection. We know that phagocytosis alone is not an effective signal for induction of NO since latex beads do not induce NO production in goldfish macrophages [63]. That the regulation of NO production in macrophages may be more primitive in teleosts, is supported by our data that MDF inhibited both A. hydrophila and LPS-mediated induction of the NO response, suggesting that MDF may exert its effects downstream of the convergent LPS A. hydrophila nitric oxide-inducing pathways. IL-4 [64] and IL-10 [65] cytokines are known to deactivate mammalian macrophages. Interleukin 10 was also shown to inhibit both the release of TNF-a by activated macrophages and MAFs derived from T cells [66,67].
Bovine TGF-β1 has been shown to inhibit ROI production of rainbow trout macrophages suggesting that this cytokine may be an important macrophage deactivator in fish [68]. The high conservation of piscine and mammalian TGF-β, the potent macrophage deactivating properties of this cytokine and the similar Mr of the biologically active forms of mammalian TGF-β 1-3 and the teleost MDF identified in this study, suggests that TGF-β may be a potential candidate for this endogenously-derived teleost MDF. Teleost homologues equivalent to the deactivating mammalian cytokines IL-4, IL-10, and IL-13 may also be potential candidates for the macrophage deactivation activities identified in this study. The precise molecular nature of MDF remains to be determined.
In recent years, many immune-related cytokine genes have been identified and characterized, which has helped in numerous studies on the expression of these genes during disease development and understanding molecular pathogenesis [69-71]. Several authors have been documented the innate as well as specific immune parameters in fish exposed to other pathogens [72-76]. The immunomodulation mechanism in fish induced by A. hydrophila is still poorly understood. The innate immune response in fish is the first line of defence against any tissue damage or pathogen interaction, which involves both fixed and mobile cells, and large number of molecules dissolved in body fluids. IL-1β is one of the crucial early response pro-inflammatory cytokines. It is enables organisms to respond to infectious insults that inducing an inflammatory cascade, along with other defensive responses. This acute phase response related genes, such as transferrin and ceruloplasmin, are induced that bind to iron and create a bacteriostatic environment by limiting the availability of iron to replicating pathogens. Some other gene products, such as lysozyme, protease inhibitors, and complement proteins may play a role in further restricting their multiplication or pathogenesis [77,78].
The changes in immune system during infection are also indicated by the up- or down-regulation of immune-related genes. Because A. hydrophila is a major problem in aquaculture industry [79,80]. The expression studies of immune-related gene in goldfish have been recently initiated our laboratory, and understanding of immune responses to A. hydrophila is limited. The present work is the first of such expression studies in goldfish for few important immune-related genes of IL-1β and IL-6. Semi-quantitative RT-PCR was performed on kidney and spleen tissues samples to identify up/down regulated mRNA expression relative to uninfected control fish and normalized to the housekeeping gene, β-actin. In our experiment, administration of bacteria intraperitoneally allowed the pathogen immediate access to the vasculature, and infected fish subsequently displayed a rapid induction of immune genes of IL-1β and IL-6.
Analyses of IL-1β mRNA expression profiles revealed evidence of up-regulation of the inflammatory response in zebrafish embryos and adults as a result of E. tarda infection [81]. Expression of c-type lysozyme was found to increase in anterior kidney, spleen, and ovary of Japanese flounder following E. tarda infection [82]. E. tarda is capable of inducing production of NO and TNFa from macrophages attributing to its high virulence [83] and extracellular products of E. tarda isolates have been noted to promote chemokinetic movement of macrophages of Nile tilapia [27].
IL-1β is a major player in immune response of fish as in mammals [84] and is a key mediator in response to microbial invasion and tissue injury that stimulate immune responses by activating lymphocytes or by inducing the release of other cytokines capable of triggering macrophages, NK cells, and lymphocytes. The macrophages are the primary source of IL-1β was found to be significantly up-regulated during E. tarda infection in rohu as noticed previously in zebrafish embryos and adults during infection [81]. The up-regulation of IL- 1β and IL-6 genes mRNA expression in the head kidney, intestine, and spleen of vaccine treated groups as suggested in Atlantic salmon vaccinated with multivalent bacterial vaccine previously [85]. Increase in IL-1β and IL-6 genes expression in the head kidney of goldfish was noticed after administration of vaccine treated groups. The increase in IL-1β gene expression indicates that induce mucus secretion and activation of macrophages as well as up-regulating the expression of a number of NF-kB-dependent genes [69]. Thus, significant upregulation of IL-1β and IL-6 gene expression in goldfish provides in this study evidence that early inflammatory immune response is stimulated upon infection with A. hydropila.
IL-1β expression can activate transcription factors such as NFkB, which has been shown to be important for iNOS transcription in fish [86]. Therefore, the present study was observed up-regulation in IL-1β gene expression may increase in iNOS expression might have helped in the production of Reactive Nitrogen Intermediates (RNIs) that may damage the pathogen at the earlier phase (data not shown). A. hydropila infection, the handling of pathogens by macrophages through production of RNI might be playing crucial role rather than iNOS-independent (more likely ROI) pathway, which is being degraded by production of antioxidant enzymes by bacterium.
Significant down-regulation of expression in the infected untreated group was noted in goldfish after infection with A. hydropila. Haugland et al. [85] observed differential expression of the two variants (types 1 and 2) in Atlantic salmon administered with an oil-based multivalent vaccine. In head kidney and spleen of formaline-killed vaccine treated group, the expression of IL-1β and IL-6 was markedly high, which showed no changes in the expression levels in control group. Similarly, the expression of IL-1β and IL-6 genes showed significant rise in the head kidney of infected goldfish after immunization with A. hydropila. This up-regulation of IL-1β and IL-6 genes might be responsible for the rise inhibition of macrophages activity of goldfish after immunization with A. hydropila as indicated in the present study. It is interesting to find that up-regulation in the expression of IL-1β and IL-6 genes were noticed vaccine treated groups whereas, down-regulation of infected untreated group. The tissue samples were collected from survived fish after challenge. Thus, up- and down-regulation of the above genes studied here might be playing important role in A. hydropila pathogenesis and survival. The present study add to the understanding of the goldfish immune responses to one important Gram-negative bacterium A. hydropila and provide a foundation of further genomic characterization of immune-related genes from this important culturable fish. Further study is needed to elucidate the function of this innate immune gene expression during A. hydropila infection, in terms of the effects on eventual survival and recovery from infection.
References
- Engelsma MY, Stet RJ, Saeij JP, Verburg-van Kemenade BM. Differential expression and haplotypic variation of two interleukin-1beta genes in the common carp (Cyprinus carpio L.). Cytokine. 2003; 22: 21-32.
- Bird S, Zou J, Kono T, Sakai M, Dijkstra JM, Secombes C. Characterisation and expression analysis of interleukin 2 (IL-2) and IL-21 homologues in the Japanese pufferfish, Fugu rubripes, following their discovery by synteny. Immunogenetics. 2005; 56: 909-923.
- Bird S, Zou J, Savan R, Kono T, Sakai M, Woo J, Secombes CJ. Characterisation and expression analysis of an interleukin 6 homologue in the Japanese pufferfish, Fugu rubripes. Dev Comp Immunol. 2005; 29: 775-789.
- Sangrador-Vegas A, Lennington JB, Smith TJ. Molecular cloning of an IL-8- like CXC chemokine and tissue factor in rainbow trout (Oncorhynchus mykiss) by use of suppression subtractive hybridization. Cytokine. 2002; 17: 66-70.
- Zou J, Clark MS, Secombes CJ. Characterisation, expression and promoter analysis of an interleukin 10 homologue in the puffer fish, Fugu rubripes. Immunogenetics. 2003; 55: 325-335.
- Wang TH, Holland JW, Bols N, Secombes CJ. Cloning and expression of the first nonmammalian interleukin-11 gene in rainbow trout Oncorhynchus mykiss. FEBS J. 2005; 272: 1136-1147.
- Robertsen B, Bergan V, Rokenes T, Larsen R, Albuquerque A. Atlantic salmon interferon genes: cloning, sequence analysis, expression, and biological activity. J Interferon Cytokine Res. 2003; 23: 601-612.
- Kono T, Zou J, Bird S, Savan R, Sakai M, Secombes CJ. Identification and expression analysis of lymphotoxin-beta like homologues in rainbow trout Oncorhynchus mykiss. Mol Immunol. 2006; 43: 1390-1401.
- Zou J, Cunningham C, Secombes CJ. The rainbow trout Oncorhynchus mykiss interleukin-1 beta gene has a different organization to mammals and undergoes incomplete splicing. Eur J Biochem. 1999; 259: 901-908.
- Hong S, Zou J, Crampe M, Peddie S, Scapigliati G, Bols N, Cunningham C, Secombes CJ. The production and bioactivity of rainbow trout (Oncorhynchus mykiss) recombinant IL-1 beta. Vet Immunol Immunopathol. 2001; 81: 1-14.
- Dinarello CA. The IL-1 family and inflammatory diseases. Clin Exp Rheumatol. 2002; 20: 1-13.
- 12. Mizel SB. The interleukins. FASEB J. 1989; 3: 2379-2388.
- Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. Journal of leukocyte biology. 2007; 81: 1-5.
- Sepulcre MP, Lopez-Castejon G, Meseguer J, Mulero V. The activation of gilthead seabream professional phagocytes by different PAMPs underlines the behavioural diversity of the main innate immune cells of bony fish. Molecular immunology. 2007; 44: 2009-2016.
- Martin SAM, Blaney SC, Houlihan DF, Secombes CJ. Transcriptome response following administration of a live bacterial vaccine in Atlantic salmon (Salmo salar). Mol Immunol. 2006; 43: 1900-1911.
- Byon JY, Ohira T, Hirono I, Aoki T. Use of a cDNA microarray to study immunity against viral hemorrhagic septicemia (VHS) in Japanese flounder (Paralichthys olivaceus) following DNA vaccination. Fish Shellfish Immunol. 2005; 18: 135-147.
- Morrison RN, Cooper GA, Koop BF, Rise ML, Bridle AR, Adams MB, Nowak BF. Transcriptome profiling the gills of amoebic gill disease (AGD)-affected Atlantic salmon (Salmo salar L): a role for tumor suppressor p53 in AGD pathogenesis? Physiol Genomics. 2006; 26: 15-34.
- Roberge C, Paez DJ, Rossignol O, Guderley H, Dodson J, Bernatchez L. Genome-wide survey of the gene expression response to saprolegniasis in Atlantic salmon. Mol Immunol. 2007; 44: 1374-1383.
- Li J, Pritchard DK, Wang X, Park DR, Bumgarner RE, Schwartz SM, Liles WC. cDNA microarray analysis reveals fundamental differences in the expression profiles of primary human monocytes, monocyte-derived macrophages, and alveolar macrophages. Journal of leukocyte biology. 2007; 81: 328-335.
- Purcell MK, Nichols KM, Winton JR, Kurath G, Thorgaard GH, Wheeler P, Hansen JD, Herwig RP, Park LK. Comprehensive gene expression profiling following DNA vaccination of rainbow trout against infectious hematopoietic necrosis virus. Mol Immunol. 2006; 43: 2089-2106.
- Mackenzie S, Iliev D, Liarte C, Koskinen H, Planas JV, Goetz FW, Molsa H, Krasnov A, Tort. Transcriptional analysis of LPSstimulated activation of trout (Oncorhynchus mykiss) monocyte/macrophage cells in primary culture treated with cortisol. Mol Immunol. 2006; 43: 1340-1348.
- McGuire K, Glass EJ. The expanding role of microarrays in the investigation of macrophage responses to pathogens. Vet Immunol Immunopathol. 2005; 105: 259-275.
- Hedges JF, Lubick KJ, Jutila MA. Gamma delta T cells respond directly to pathogen-associated molecular patterns. J Immunol. 2005; 174: 6045-6053.
- Wang JP, Rought SE, Corbeil J, Guiney DG. Gene expression profiling detects patterns of human macrophage responses following Mycobacterium tuberculosis infection. FEMS Immunol Med Microbiol. 2003; 39: 163-172.
- Ehrt S, Schnappinger D, Bekiranov S, Drenkow J, Shi S, Gingeras TR, Gaasterland T, Schoolnik G, Nathan C. Reprogramming of the macrophage transcriptome in response to interferongamma and Mycobacterium tuberculosis: signalling roles of nitric oxide synthase-2 and phagocyte oxidase. J Exp Med. 2001; 194: 1123-1140.
- Jensen K, Talbot R, Paxton E, Waddington D, Glass EJ. Development and validation of a bovine macrophage specific cDNA microarray. BMC Genomics. 2006; 7: 224.
- Koskinen H, Pehkonen P, Vehniainen E, Krasnov A, Rexroad C, Afanasyev S, Molsa H, Oikari A. Response of rainbow trout transcriptome to model chemical contaminants. Biochemical and biophysical research communications. 2004; 320: 745-753.
- Krasnov A, Koskinen H, Pehkonen P, Rexroad CE 3rd, Afanasyev S, Molsa H. Gene expression in the brain and kidney of rainbow trout in response to handling stress. BMC genomics. 2005; 6: 3.
- MacKenzie S, Iliev D, Liarte C, Koskinen H, Planas JV, Goetz FW, Molsa H, Krasnov A, Tort L. Transcriptional analysis of LPS stimulated activation of trout (Oncorhynchus mykiss) monocyte/macrophage cells in primary culture treated with cortisol. Molecular immunology. 2006; 43: 1340-1348.
- Iliev DB, Liarte CQ, MacKenzie S, Goetz FW. Activation of rainbow trout (Oncorhynchus mykiss) mononuclear phagocytes by different pathogen associated molecular pattern (PAMP) bearing agents. Mol Immunology. 2005; 42: 1215-1223.
- Byon JY, Ohira T, Hirono I, Aoki T. Use of a cDNA microarray to study immunity against viral hemorrhagic septicemia (VHS) in Japanese flounder (Paralichthys olivaceus) following DNA vaccination. Fish Shellfish Immunol. 2005; 18: 135-147.
- Ewart KV, Belanger JC, Williams J, Karakach T, Penny S, Tsoi SC, Richards RC, Douglas SE. Identification of genes differentially expressed in Atlantic salmon (Salmo salar) in response to infection by Aeromonas salmonicida using cDNA microarray technology. Dev Comp Immunol. 2005; 29: 333-347.
- Kasahara M, Naruse K, Sasaki S, et all. The medaka draft genome and insights into vertebrate genome evolution. Nature. 2007; 447: 714-719.
- Rise ML, Jones SR, Brown GD, von Schalburg KR, Davidson WS, Koop BF. Microarray analyses identify molecular biomarkers of Atlantic salmon macrophage and hematopoietic kidney response to Piscirickettsia salmonis infection. Physiological genomics. 2004; 20: 21-35.
- Joerink M, Ribeiro CM, Stet RJ, Hermsen T, Savelkoul HF, Wiegertjes GF. Head kidney-derived macrophages of common carp (Cyprinus carpio L.) show plasticity and functional polarization upon differential stimulation. J Immunol. 2006; 177: 61-69.
- Bishop RE. Fundamentals of endotoxin structure and function. Contributions to microbiology. 2005; 12: 1-27.
- Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Ann Rev Biochem. 2002; 71: 635-700.
- Virelizier JL, Arenzana-Seisdedos F. Immunological functions of macrophages and their regulation by interferons. Med Biol. 1985; 63: 149-159.
- Nathan C, Hibbs Jr JB. Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr Opin Immunol. 1991; 3: 65-70.
- Green SJ, Nacy CA. Antimicrobial and immunopathological effects of cytokine-induced nitric oxide synthesis. Curr Opin Infect Dis. 1993; 6: 384- 390.
- Karupiah G, Xie-Q-W, Buller ML, Nathan CA, Duarte C, MacMicking JD. Inhibition of viral replication by interferon-gamma-induced nitric oxide synthase. Science. 1993; 261: 1445-1449.
- Belosevic M, Davis CE, Meltzer MS, Nacy CA. Regulation of activated macrophage antimicrobial activities: identification of lymphokines that induce resistance to infection with Leishmania major. J Immunol. 1988; 141: 890- 896.
- Lorsbach RB, Murphy WJ, Lowenstein CJ, Snyder SH, Russell SW. Expression of the nitric oxide synthase gene in mouse macrophages activated for tumor cell killing: molecular basis for the synergy between interferongamma and lipopolysaccharide. J Biol Chem. 1993; 268: 1908-1913.
- Neumann NF, Fagan D, Belosevic M. Macrophage activating factor(s) secreted by mitogen stimulated goldfish kidney leukocytes synergize with bacterial lipopolysaccharide to induce nitric oxide production in teleost macrophages. Dev Comp Immunol. 1995; 19: 473-482.
- Marletta MA, Yoon PS, Ivengar R, Leaf CD, Wishnok JS. Macrophage oxidation of L-arginine: nitric oxide is an intermediate. Bioch. 1988; 27: 8706- 8711.
- Dõâallwssio G, DiDonato A, Jaffe K, Maldonado H, Zalbala NA. Arginine and memory consolidation in praying mantis. J Comp Physiol. 1982; A47: 231- 234.
- Martinez A, Riveros-Moreno V, Polak JM, Moncada S, Sesma P. Nitric oxide (NO) synthase immunoreactivity in the starfish Marthasteria glacialis. Cell Tiss Res. 1994; 275: 599-603.
- Wang R, Nemann NF, Shen Q, Belosevic M. Establishment and characterization of a macrophage cell line from the goldfish. Fish Shellfish Immunol. 1995; 5: 329-346.
- Neumann NF, Belosevic M. Antimicrobial mechanisms of activated macrophages and their regulation by cytokines. Adv Struct Biol. 1995; 4: 233- 254.
- Rath R K. Freshwater Aquaculture. Scientific Publishers, Jodhpur, India. 1993; 282-212.
- Karunasagar I, Ali A, Otta SK, Karunasagar I. Immunization with bacterial antigens: infections with motile Aermonas. Gudding R, Lillehaug A, Midtlyng PJ, eds. Fish Vaccinology. Dev Biol Stands Basel Karger. 1997; 90: 1-7.
- Actor JK, Hwang S, Olsen M, Zimecki M, Hunter RL Jr, Kruzel ML. Lactoferrin immunomodulation of DTH response in mice. Int Immunopharmacol. 2002; 2: 475-486.
- Sakai M. Current research status of fish immunostimulants. Aquaculture. 1999; 172: 63-92.
- Raa J. The use of immune-stimulants in fish and shellfish feeds. Cruz –Suarez LE, Ricque-Marie D, Tapia-Salazar M, Olvera-Novoa MS, Civera-Cerecedo R, editors. Avances en Nutricion Acuicola V. Memorias del V Simposium Internacional de Nutricion Acuicola. Merida, Yucatan, Mexico. 2000; 47-56.
- Joseph SW, Carnahan A. The isolation, identification, and systematics of the motile Aeromonas species. Annu Rev Fish Dis. 1994; 4: 315-343.
- Davis JF, Hayasaka SS. Pathogenic bacteria associated with cultured American eels Anguilla rostrata Le Sueur. J Fish Biol. 1983; 23: 557-564.
- Harikrishnan R, Balasundaram C, Heo MS. Effect of chemotherapy, vaccination and immunomodulation in goldfish, Carassius auratus against Aeromonas hydrophila. Dis Aquat Org. 2009; 88: 45-54.
- Akhlaghi M, Munday BL, Whittington RJ. Comparison and active immunization of fish against streptococcosis (enterococcosis). J Fish Dis. 1996; 19: 251- 258.
- Eko FO, Szostak MP, Wanner G, Lubitz W. Production of Vibrio cholerae ghosts (VCG) by expression of a cloned phage lysis gene: potential for vaccine development. Vaccine. 1994; 12: 1231-1237.
- Amend D. Potency testing of fish vaccines. Dev Biol Stand. 1981; 49: 447- 454.
- Neumann NF, Barreda D, Belosevic M. Production of a macrophage growth factor(s) by a goldfish macrophage cell line and macrophages derived from goldfish kidney leukocytes. Dev Comp Immunol. 1998; 4: 417-432.
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite and 15-nitrate in biological fluids. Anal Biochem. 1982; 126: 131-138.
- Neumann NF, Belosevic M. Antimicrobial mechanisms of activated macrophages and their regulation by cytokines. Adv Struct Biol. 1995; 4: 233- 254.
- Liew FY, Li Y, Severn A, Millott S, Schmidt J, Salter M, Moncada S. A possible novel pathway of regulation by murine T helper type-2 (Th2) cells of a Th1 cell activity via the modulation of the induction of nitric oxide synthase on macrophages. Eur J Immunol. 1991; 21: 2489-2494.
- Stenger S, Thuring H, Rollonghoff M, Bogdan C. Tissue expression of inducible nitric oxide synthase is closely associated with resistance to infection with Leishmania major. J Exp Med. 1994; 180: 783-793.
- Bogdan C, Vodovotz Y, Nathan CA. Macrophage deactivation by interleukin-10. J Exp Med. 1991; 174: 1549-1555.
- Fiorentino DF, Zlotnik A, Viera P, Mosmann TR, Howard M, Moore KW, O’Garra A. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J Immunol. 146: 3444-3451.
- Jang SI, Hardie LJ, Secombes CJ. Effects of transforming growth factor b1 on rainbow trout Oncorhynchis mykiss macrophage respiratory burst activity. Dev Comp Immunol. 1994; 4: 315-323.
- Lindenstrom T, Secombes CJ, Buchmann K. Expression of immune response genes in rainbow trout skin induced by Gyrodactylus derjavini infections. Vet Immunol Immunopathol. 2004; 97: 137-148.
- Purcell MK, Kurath G, Garver KA, Herwig RP, Winton JR. Quantitative expression profiling of immune response genes in rainbow trout following Infectious Haematopoietic Necrosis Virus (IHNV) infection or DNA vaccination. Fish Shellfish Immunol. 2004; 17: 447-462.
- Sigh J, Lindenstrom T, Buchmann K. The parasitic ciliate Ichthyophthirius multifiliis induces expression of immune relevant genes in rainbow trout, Oncorhynchus mykiss (Walbaum). J Fish Dis. 2004; 27: 409-417.
- Roed KH, Brun E, Larsen HJ, Refstie T. Genetic variation in serum haemolytic activity in Atlantic salmon (Salmo salar L.). J Fish Biol. 1992; 40: 739-750.
- Roed KH, Larsen HJS, Linder RD, Refstie T. Genetic variation in lysozyme activity in rainbow trout (Oncorhynchus mykiss). Aquaculture. 1993; 109: 237-244.
- Roed KH, Fevolden S-E, Fjalestad KT. Disease resistance and immune characteristics in rainbow trout (Oncorhynchus mykiss) selected for lysozyme activity. Aquaculture. 2002; 209: 91-101.
- Sarder MRI, Thompson KD, Penman DJ, McAndrew BJ. Immune responses in Nile tilapia (Oreochromis niloticus L.) clones: I. Non-specific responses. Dev Comp Immunol. 2001; 25: 37-46.
- Gross KA, Powell MD, Butler R, Morrison RN, Nowak BF. Changes in the innate immune response of Atlantic salmon, Salar salar L., exposed to experimental infection with Neoparamoeba sp. J Fish Dis. 2005; 28: 293-299.
- Ellis AE. Innate host defense mechanisms of fish against viruses and bacteria. Dev Comp Immunol. 2001; 25: 827-839.
- Magnadottir B. Innate immunity of fish (overview). Fish Shellfish Immunol. 2006; 20: 137-151.
- Swain P, Behura A, Dash S, Nayak SK. Serum antibody response of Indian major carp, Labeo rohita to three species of pathogenic bacteria; Aeromonas hydrophila, Edwardsiella tarda and Pseudomonas fluorescens. Vet Immunol Immunopathol. 2007; 117: 137-141.
- Steel DM, Whitehead AS. The major acute phase reactants: C-reactive protein, serum amyloid P and serum amyloid A protein. Immunol Today. 1994; 15: 81-88.
- Pressley ME, Phelan III PE, Witten PE, Mellon MT, Kim CH. Pathogenesis and inflammatory response to Edwardsialla tarda infection in the zebrafish. Dev Comp Immunol. 2005; 29: 501-513.
- Hikima J, Hirono I, Aoki T. Characterization and expression of c-type lysozyme cDNA from Japanese flounder (Paralichthys olivaceus). Mol Mar Biol Biotechnol. 1997; 6: 339-344.
- Ishibe K, Yamanishi T, Wang Y, Osatomi K, Hara K, Kanai K, et al. Comparative analysis of the production of Nitric Oxide (NO) and tumor necrosis factor- a (TNF-a) from macrophages exposed to high virulent and low virulent strains of Edwardsiella tarda. Fish Shellfish Immunol. 2009; 27: 386-389.
- Secombes CJ. The nonspecific immune system: cellular defences. In: Iwama G, Nakanishi N, editors. The fish immune system: organism, pathogen and environment. San Diego: Academic Press Inc. 1996; 63-103.
- Haugland O, Torgersen J, Syed M, Evensen O. Expression profiles of inflammatory and immune-related genes in Atlantic salmon (Salmo salar L.) at early time post vaccination. Vaccine. 2005; 23: 5488-5499.
- Saeij JPJ, Stet RJM, Groeneveld A, Verburg-van Kemenade LBM, van Muiswinkel WB, Wiegertjes GF. Molecular and functional characterization of a fish inducible-type nitric oxide synthase. Immunogenetics. 2000; 51: 339- 346.