
Case Report
Ann Carcinog. 2019; 4(1): 1016.
An Unusual Adrenal Tumor not to be Ignored!
Tbouda M1*, Toufga Z2, Belhabib S1, Zouaidia F1, Znati K1, Jahid A1 and Bernoussi Z1
¹Department of Anatomical Pathology, Ibn Sina Hospital, Morocco
²Department of Radiology, Ibn Sina Hospital, Morocco
*Corresponding author: Tbouda M, Department of Anatomical Pathology, Ibn Sina Hospital, Faculty of Medicine and Pharmacy, Mohammed V University, Hôpital Militaire Mohamed V, Rabat, Morocco
Received: February 19, 2019; Accepted: March 22, 2019; Published: March 29, 2019
Abstract
Adrenal tumors are dominated by epithelial tumors. Mesenchymal tumors are rare. Among these tumors is Leiomyosarcoma (LS), which is extremely rare. It originates from the muscle cells of the muscular is of the adrenal veins. Immunocompromised individuals are the most susceptible to this type of tumor. We report a case of LS in a 17-year-old patient presenting for an abdominal mass in a context of general deterioration. Imaging had suspected the diagnosis of a malignant adrenal tumor. Laparoscopic surgical resection was successfully performed. Clinical and radiological control was performed with a follow-up of 12 months.
A rare and aggressive tumor that most often requires radical surgery sometimes extended to neighboring organs. Chemotherapy and radiotherapy are indicated in case of contraindication to surgery.
Keywords: Adrenal; Tumor; Leiomyosarcoma; Histology
Abbreviations
LS: Leiomyosarcoma; CT: Tomography
Introduction
Adrenal leiomyosarcoma is a rare tumor. Its frequency does not exceed 50 cases reported in the literature [1]. Its etiology is unknown, however, some authors estimate its high incidence in the population of individuals infected with HIV or Epstein-Barr virus [2]. It is an aggressive tumor that only radical surgery can improve survival. Chemotherapy and radiotherapy only indicated in case of unresectable and metastatic tumor [2]. We report here a case of leiomyosarcoma in a young subject, while describing the diagnostic circumstances and the therapeutic approach, while going through a review of the literature.
Case Report
This is a 49-year-old patient with no particular history who consulted for a progressive increase in the abdomen, in a context of general deterioration. The clinical examination finds a conscientious, afebrile patient with impaired facies. The abdominal examination shows a mass of the right flank, deep, painless. An ultrasound was performed showing a hyperechoic mass with hypoechoic areas at the right renal area. Completed by Computed Tomography (CT), the mass appeared poorly limited, heterogeneously enhanced after injection of contrast medium including vascular structures with ipsilateral renal pedicle extension and perirenal fat infiltration measuring 22×7 cm (Figure 1,2). The patient was hospitalized in the urology ward. A biological tests was carried out without particularities and including an HIV serology which was negative. Surgical resection of the tumor laparotomically has been successfully performed.
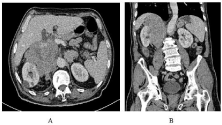
Figure 1: Axial (A) and coronal (B) CT of abdominal CT with contrast injection
showing a tumor of the right adrenal area poorly limited, heterogeneously
enhanced after injection of contrast medium.
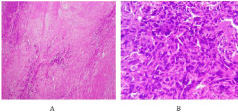
Figure 2: Histological aspect at low magnification (HE×100) (A) showing a
partially necrotic fusocellular tumor proliferation. And at medium magnification
(HE×200) (B): note atypies and mitoses.
The tumor extracted weighed 1350 g. Several samples were taken and examined under the microscope. Histologically, this is a partially necrotic fusocellular tumor proliferation made of long bundles of cells with marked atypia (Figure 3). An immunohistochemical complement realized showing a positive H-caldesmon, smooth muscle and Desmin antibodies. CD34, CK AE1-AE3 and myogenin antibodies did not show tumor cell labeling.
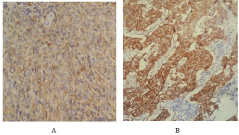
Figure 3: Positive labeling of tumor cells with the Desmine antibody (A) and
H-caldesmone antibody (B).
Discussion
Adrenal mesenchymal tumors are rare. Only a few cases have been reported in the literature [1]. The age of patients varies between 30 and 60 years [2]. More than 20 years have been reported in the literature, our 49-year-old patient. Men and women are equally affected by this disease [1]. Some authors thought that its frequency was high in HIV carriers or infected with Epstein-Barr virus (EBV) [1]. Our patient has a negative HIV serology, which opens the door to the pathogenesis still poorly elucidated of this rare adrenal pathology. These tumors are generally large and according to the literature, adrenal tumors greater than 3 cm, are suspected of malignancy [4].
Abdominal ultrasonography usually shows a lobulated, heterogeneous retro-peritoneal tumor [5], which detects lesions larger than 3 cm [6]. The abdominal CT after injection of contrast medium is the reference examination, allows to specify the nature of the mass, often polylobed contours, with a heterogeneous enhancement after injection of contrast medium. It determines its location, its relations with neighboring bodies, and thus allows a loco-regional resectability and expansion assessment [5,6]. Magnetic Resonance Imaging (MRI) does not provide additional elements compared to newer CT generations [6-8]. According to some authors, the combination of CT, MRI and angiography is useful for the diagnosis and for the pre-therapeutic assessment of adrenal leiomyosarcomas [9]. In our patient, the abdominal CT scan was sufficient to perform a complete evaluation showing a localized adrenal tumor with vascular invasion of the renal hilum.
Diagnostic confirmation is provided by the histological analysis performed most often on the operative specimen, rarely on scanned-guided biopsy material. It is an obviously malignant tumor proliferation that produces large cell bundles. Tumor cells are elongated with abundant eosinophilic cytoplasm and nuclei showing marked atypia with many mitotic figures. This histological analysis must be completed by an immunohistochemical study. This tumor shows positivity for smooth muscle markers, particularly smooth muscle actin (AML), H-caldesmone and desmin [10].
Like most malignant tumors, complete resection with healthy margins is the only effective therapy that prolongs patient survival. According to Mencoboni et al. [11], postoperative radiotherapy is recommended for locally advanced tumors. The effectiveness of chemotherapy is poorly defined and very limited. Its indication is limited to inoperable tumors and incompletely resected tumors or metastatic tumors [1].
Conclusion
Adrenal tumors are dominated by epithelial tumors. Mesenchymal tumors are rare. Among these tumors is Leiomyosarcoma, which is extremely rare. It is an aggressive tumor that most often requires radical surgery sometimes extended to neighboring organs. Chemotherapy and radiotherapy are indicated in case of contraindication to surgery.
References
- Dekoua A, N’Dahb J.K, Kouamea B, Kohouc L, Abroulaye F, Gowe E, et al. Léiomyosarcome primitif de la surrénale chez une noire africaine : aspects diagnostiques et thérapeutiques. EM-consulte. 2013; 01: 14.
- Lujan MG, Hoang MP. Pleomorphic leiomyosarcoma of the adrenal gland. Arch Pathol Lab Med. 2003; 9: 32-35.
- Soufi M, Mohsine R, Chenna M, Mouaquit O, El Malki HO, Ifrine L, et al. Léiomyosarcome primitif de la surrénale. J Afr Cancer. 2009; 1: 168-171.
- Welsh SJ, Khan S. Radiological localizing techniques in adrenal tumors. Minerva Endocrinol. 2009; 34: 161-169.
- Hammoune N, El Guendouz F, Elhaddad S, Latib R, Chami I, Boujida I, et al. Léiomyosarcome de la veine cave inférieure: un cas clinique. J Afr Cancer. 2015; 20: 238.
- Yung-Liang W. Ultrasonography of the Adrenal Gland. J Med Ultrasound. 2007; 15: 213-227.
- Soury P, Lepechou C, Guinebretiere JM, Laurian C. Léiomyoscarcomes de la veine cave inférieure. EMC-Cardiologie Angéiologie. 2005; 2: 90-96.
- Ben Hassouna J, Bouzaiene H, Chargui R, Ben Bachouche W, Khomsi F, Habib Mtaalah M, et al. Léiomyosarcome de la veine cave inférieure sousrénale. J Chir. 2006; 143: 325-327.
- Lee CW, Tsang YM, Liu KL. Primary adrenal leiomyosarcoma. Abdom Imaging. 2006; 31: 123-124.
- Carvalho JC, Thomas DG, Lucas DR. Cluster analysis of immunohistochemical markers in leiomyosarcoma delineates specific anatomic and gender subgroups. Cancer. 2009; 115: 4186-41895.
- Mencoboni M, Bergaglio M, Truini M, Varaldo M. Primary adrenal leiomyosarcoma: a case report and literature review.Clin Med: Oncol. 2008; 2: 353-356.