
Review Article
Austin J Clin Immunol. 2015; 2(1):1025.
Intratumoral Immune Landscape: Immunogenicity to Tolerogenicity
Abir K Panda, Sayantan Bose, Sreeparna Chakraborty, Kirti Kajal and Gaurisankar Sa*
Division of Molecular Medicine, Bose Institute, India
*Corresponding author: Gaurisankar Sa, Division of Molecular Medicine, Bose Institute, P-1/12, CIT Scheme VII M, Kolkata-700054, India
Received: April 13, 2015; Accepted: September 05, 2015; Published: September 10, 2015
Abstract
Immune system possesses distinct innate (less specific) and adaptive (more specific) branches which act in a collaborative way to eliminate cancer from the host. In spite of the presence of immune response, tumors develop in the body spontaneously through different immune escape strategies. During the progression of cancer, immune cells become paralyzed and altered. In tumor microenvironment both innate (macrophage and NK cells) and adaptive (CTLs and effector T cells) immune cells are unable to recognize and induce specific effector response against cancer to eradicate it. Tumor cells release different types of chemokines, cytokines, growth factors that can modulate immune cells to become tolerogenic and allow tumor cells to grow rapidly without any restriction. Immune cells also cannot discriminate the tumor antigens as they are concealed in stroma and are also less immunogenic. The immune cells thus become dormant and effective immune responses against tumors could not be elicited. Tumor cells exploit the plethora of immunosuppressive mechanisms which include abnormalities of antigen processing and presentation, induction of negative co-stimulatory signals that helps to establish tumor immune evasion. In addition, infiltration of T-regulatory cells, immature and tolerogenic Dendritic Cells (DCs), tumor-associated macrophages, and myeloid-derived stromal cells foster suppressive, tolerogenic condition. The understanding of different immune evasion mechanisms will help to design effective immunotherapies to overcome tolerogenic condition and elicit tumor regression.
Keywords: Immune cell dysfunction; Immunogenicity; Tolerogenicity; Tumor immune evasion; Tumor micro-environment
Introduction
The innate and adaptive immune system work together to identify foreign pathogens as well as cancerous outgrowths in the host body and induces effective immune responses to eliminate them. But over the decades it has been a mystery, how tumor develops in the host in spite of the immune system’s potential to recognize and destroy them. In 1863 Rudolf Virchow first suggested that there was a functional relation between leukocyte infiltration and malignant growth. In 1957 Brunet and Thomas postulated the immune surveillance theory which stated that the immune system can counter attack developing tumor in a host [1]. However, this concept remained debatable due to the lack of experimental evidence. After a long period in 2003, new evidence indicated that immune system can eliminate tumors through immune surveillance [2,3]. During the process of tumor development, the tumor microenvironment, which is composed of tumor cells, immune cells, extracellular matrix, and stromal cells, produces certain key factors that helps in fostering tumor growth, proliferation and also promote metastasis. During tumor progression, modifications occur in certain signaling pathways of immune system that induces tumor immune tolerance and subsequently escape tumor immunity. In 2003 Robert Schreiber put forward the well accepted immune editing hypothesis which composed of three distinct phases: (i) Elimination, (ii) Equilibrium and (iii) Escape. In elimination or immune surveillance phase immune system can recognize and eliminate developing tumor thus protecting host against tumor. In equilibrium phase tumor cells and immune cells may enter into a dynamic equilibrium those results in tumor persistence. In escape phase tumor variants that escape from immune selection process of equilibrium stages develop into clinically apparent, highly metastatic and invasive tumors by avoiding the immune responses [4,5]. In tumor microenvironment the tumor cells employ certain strategies to escape immune response by modulating the immune cells so that they cannot recognize and eliminate them. When tumors become highly metastatic and invasive, the immune system becomes paralyzed and the improper immune response favors tumor progression. In this review we will discuss the tumor immune escape strategies and the role of immune cells in tumor immune evasion.
Tumor immune escape strategies
In tumor microenvironment tumor cells execute various strategies to evade the immune system and establishes immunogenic to tolerogenic environment.
Tumor-associated antigens (TAA) shows low immunogenicity
It is a well established that Tumor cell expressing Antigens (TAA) are not specifically neo-antigens that are exclusively expressed in tumor cells; rather they are tissue differentiation antigens also expressed in certain normal healthy cells [6]. Hence this creates the problem of generation of immune response against such tumor antigens. Tumor-associated antigens in early metastatic stage are embedded within the solid tumor [7]. The stromal cells near tumor site prevent efficient release of TAA in draining lymph nodes [8]. In late metastatic phase, efficient TAA release induces effective immunity; however immune tolerance to TAA develops by this stage and disables the function of APCs and other T-effector cells [9]. Like chronic inflammation, human tumorigenesis is a slow process that hampers early activation of NK cell and reshapes TAA specific priming to T cell specific tolerogenic responses [10] (Figure 1).
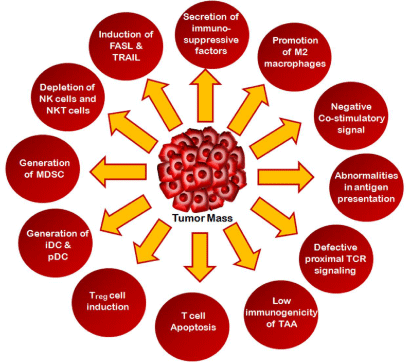
Figure 1: Major strategies adopted by tumor cells for immune evasion.
Tumor cells adopt several strategies to avoid the continuous surveillance by the immune system. The major processes by which tumor cells escape from the immune attack are outlined here.
T cell tolerance, anergy and apoptosis induces immunosuppression
To protect any kind of antigenic assault as well as cancer, the immune system possesses long term specific adaptive immune responses where T cells play a major role. Despite major advances in characterization of T cells in other infectious disease, the role of T-effector cells in cancer is not well understood. The phenotype and functionality of T-effector cells are dramatically modulated by the tumor microenvironment [11]. Tc cells which are present in tumor microenvironment recognize tumor antigens in association with MHC1 through the T cell receptor. CD8+ Tc cells kill tumor cells in a MHC1 restricted manner by perforin/granzyme, FASL- FAS or TNFα mediated TRAIL ligand based apoptosis [12]. Despite the specificity of CD8+ T cytotoxic (Tc) cells in their cell-mediated killing, many tumors express low levels of class I MHC molecule thereby perturbing such effector responses. CD4+ Th1 cells on the other hand enhances and supports the immune system by secreting cytokines such as IFNγ, TNFα and IL2 that stimulate the development of Tc cells and also orchestrates the activation and recruitment of innate immune cells [13]. Activated Th1 cells also recognize IL10-producing tumor-promoting M2 macrophages and convert those into IFNγ- producing tumor-inhibiting M1 macrophages [14]. The M1 and M2 macrophage functions are directly correlated with Th1 and Th2 cell responses. Th2 cells release IL4 which may block neo-angiogenesis indirectly [15]. Although both CD4+ Th and CD8+ Tc cells have immunogenic functions, but in tumor microenvironment these cells become tolerogenic and unresponsive towards the tumor cells [15,16]. These tolerogenic T cells show low to intermediate levels of TCR affinity against MHC restricted antigen recognition [17,18]. In tumor condition tolerogenic antigen presenting cells such as dendritic cells cross-present the tumor antigens but they can rarely activate or weakly stimulate self/tumor-specific T cells [19]. Tumor exposed T-cells also exhibit anergy and exhaustion that allow them to execute hyporesponsiveness. T cell anergy is characterized as inability of T cells to produce IL2 upon re-stimulation and allowing cell cycle arrest in antigen-independent tumor environment [20]. In anergic state naïve T cells undergo low co-stimulatory and high inhibitory stimulation. Imbalance between this low co-stimulatory and high inhibitory signaling causes improper downstream TCR-mediated signaling through diminished protein levels and dys-regulated phosphorylation [20,21]. T cell exhaustion on the other side is characterized by progressive loss of proliferation and effector cytokine production resulting in T cell apoptosis [22]. In different human tumor models it was observed that B7H1/PD1 signaling predominated in exhausted CD8+ T cell and PD1 acts as exhausted T cell marker [23]. T cells also shows senescence properties by shortening telomere, low CD28 expression and accumulation of cell cycle control proteins such as p16, p21 or p53 that inhibit cell proliferation [24,25]. Senescent T cell also possesses defective killing properties and induces inhibitory regulatory function in tumor milieu [26,27]. Memory CD8+ T cells and naïve CD4+ T cells also possesses stem cell like properties and may differentiate into different subsets of T-effector cells in varying circumstances of the tumor microenvironment [28]. In murine model it was noticed that CD44loCD62Lhi memory CD8 T cell expresses stem cell antigen-1, BCL2 and IL2 receptor and possesses self-renewal and multipotent capacity [29]. The anergy, exhaustion, senescence or stemness properties in T cells are major inducers of tumor immune evasion.
Abnormalities in antigen presentation and TCR signaling
In contrast to tumor associated antigen discrimination, tumor cells also avoid T cell-mediated immune response by impairment of antigen presenting machinery [30]. Constant generation of tumor variants by high frequency mutation can result in escape from the regime of T cell attack until some antigens are presented by stromal cells and cross reacted with CTLs for their elimination [31]. Downregulation of the antigen presenting machinery is one of the most common strategies exploited by tumor cells to avoid T cell immune response. Frequent mutation of β2-microglobulin and MHC-Iα chain decreases the expression MHC-I complex as well as selective loss of HLA alleles [32,33]. Mutation of Transporter Associated Proteins (TAPs) with antigen processing and components of immune proteasome complex LMP2 and LMP7 also induce immune evasion in cancer [30,32]. In metastatic stage Tumor Infiltrating Lymphocytes (TILs) exhibit decreased levels of CD3ζ chain and p56LCK and p59FYN tyrosine kinase which play a crucial role in TCR signaling that leads to T cell activation [31,34]. Recent study also shows that impairment of proximal TCR signaling inhibits CTL lytic function and restricts effector immune response in advance stages of cancer [35].
Negative co-stimulatory signals
Immune checkpoints that induce negative co-stimulatory signal also play crucial roles in tumor immunosuppression. Cytotoxic T Lymphocytes-4 (CTLA4) which is exclusively expressed on activated T cells primarily counteract the activities of CD28 and induce negative inhibitory signals to restrict T cell activation [36]. After being engaged by TCR and antigen, CD28 strongly amplifies T cell activation signaling. CD28 and CTLA4 both can interact with same identical ligands CD80 and CD86 [37,38]. CTLA4 has higher affinity for both of the ligands thus outcompeting CD28 binding with CD80 and CD86. CTLA4 binding inhibits protein phosphatase SHP2 and kinase PP2A that are crucial for T cell activation in tumor milieu [37,39,40]. CTLA4 also sequesters CD80 and CD86 from CD28 engagement and remove them from antigen presenting cell surface [41]. CTLA4 is also expressed by CD4+CD25+FoxP3+ Treg cells and accelerate suppressor function in tumor microenvironment [42]. Like CTLA4 another immune checkpoint that contributes to tumor immune escape involves the interaction between PD1 and Program Death receptor Ligand-1 and -2 (PDL1 and PDL2). PDL1 and PDL2 are also known as B7H1 and B7DC respectively. PD1 inhibits the kinase that activates phosphatase SHP2 during T cell activation [43,44]. PD1 can be expressed by Tumor Infiltrating Lymphocytes (TILs) in different tumors including tumor induced Treg and CTLs [45-47]. Two distinct mechanisms for the regulation of PDL1 by tumor have emerged: (i) innate immune resistance and (ii) adaptive immune responses. In some cancer it has been observed that generation of constitutive oncogenic signal in tumor cell induces the expression of PDL1. The expression of PD1 on glioblastoma increases with subsequent deletion of PTEN that associates with PI3K-AKT signaling [48] (Figure 2).
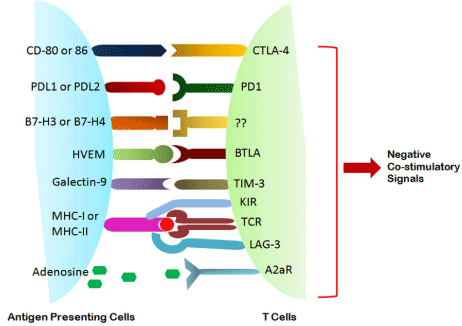
Figure 2: Negative Co-stimulatory signals between APC and Tcells in tumor
microenvironment.
Various co-stimulatory interactions between T cells and Antigen Presenting Cells (APC) are required in addition to TCR stimulation from proper T cell activation or clonal proliferation. On the contrary, in tumor milieu the different negative co-stimulatory molecular interaction between T cell and Antigen Presenting Cells (APC) drive them to clonal energy. Some of the above mentioned negative costimulatory molecules expressed on T cells are CTLA4, PD1, TIM- 3, BTLA which correspondingly interacts with CD80/86, PDL1/2, galectin-9, HVEM of APC.
Constitutive Anaplastic Lymphoma Kinase (ALK) signaling in lung cancer drives PDL1 expression through STAT3 signaling [49]. In adaptive immune resistance mechanisms PDL1 expression emerges in response to adaptation to endogenous tumor specific immune response. Expression of PDL1 occurs predominantly in tumor cells in response to PD1 specific T cells or other immune cells releasing IFNγ [50-52]. In addition to lymphocyte checkpoint inhibitory receptors B7H3 and B7H4 ligands also have inhibitory roles in cancer [53]. Lymphocyte Activation Gene-3 (LAG-3) is one of the major immune-checkpoint receptors predominantly expressed in Treg cells as well as other exhausted and anergic T cells that induce tolerance in tumor specific CD8+ T cells [54]. Galectin-9 is upregulated in various cancers including breast cancer that interacts with TIM3 ligand of CD4+IFNγ+ Th1 and CD8+ CTL and induces inhibitory signals leading to T cell anergy and tolerance [55]. Herpes Virus Entry Mediator (HVEM) ligand expressed in melanoma and tumor associated endothelial cells interact with BTLA4 (B and T cell ligand attenuator-4) in virus infected CD8+ T cells and restricts antitumor immune response [56]. There are other several types of inhibitory receptors that allow such negative co-stimulatory signals that lead to tumor immune evasion.
Immunosuppressive factors
There are various tumor derived factors that contribute to the immunosuppressive network prevalent in tumor microenvironments. TGFβ is the pleiotropic cytokine that inhibits T cell activation, induces differentiation, proliferation as well as maturation of dendritic cells and macrophages. TGFβ is secreted by tumor cells and different immune cells such as Tregs, Tumor-Associated Macrophages (TAM) and NKT cells [57]. TGFβ specifically acts on CTLs to repress the transcription of perforin, granzyme and IFNγ that are collectively involved in tumor immune responses [58]. TGFβ also promotes the proliferation of macrophages and fibroblasts that secrete some angiogenic and anti-apoptotic factors like VEGF and cyclooxyginase-2 [59]. The angiogenic factor VEGF induces immature myeloid cells that further transform into Tumor-induced immature Dendritic Cells (TiDC) and TAM in the presence of other immunosuppressive factors such as PGE2, IL10 [60]. The increased levels of PGE2, IL10 and TGFβ inhibited MHC-I / MHC-II and TAP 1/TAP2 expression on DCs and convert them into tolerogenic DCs that are unable to induce CTL-mediated immune responses [61,62]. Immunoregulatory enzyme Indole-amine 2,3 Dioxygenase (IDO) contributes to the establishment of immune tolerance by catalyzing tryptophan breakdown into kynurenine pathway metabolites [63]. Thus IDO depletes local tryptophan concentration and increased downstream metabolites confer apoptosis and anergy of T cell as well as induce Treg cells, TiDC and TAM [63]. Tumor derived gangliosides inhibits T cell activation, alter NK cell cytotoxic activities and restricts MHC-I and II mediated antigen presentation [64,65]. Gangliosides sequester IL2 and prevent it to bind to its receptor. Thus perturbed IL2 signaling pathway inhibits T cell proliferation [66]. In hypoxic tumor microenvironment macrophages release hydroxide, TGFβ, IL4 and IL10 that contribute to immune escape (Figure 3).
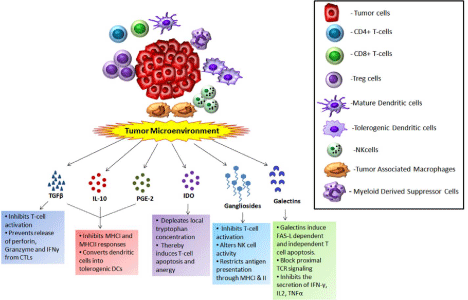
Figure 3: Different immunosuppressive factors and their interaction with immune cells in the tumor microenvironment.
The tumor microenvironment consists of higher number of tolerogenic cells like Tregs, TAM and tolerogenic DC and lower numbers of T-effector cells. The tumor mass along with these tolerogenic immune cells secrete a number of immunomodulatory factors like TGFβ, IL10, PGE2, gangliosides, galectins etc. These molecules foster a tolerogenic environment and block effector immune responses against the tumor through various mechanisms.
Soluble FAS Ligand (sFASL) and soluble MHC class I–related Chain A gene (sMICA) promotes immune evasion by inhibiting Fas and NKG2D-mediated killing of immune cells respectively [67]. Soluble Phosphotidylserine (sPS) also induces anti-inflammatory responses to TAM that secrete TGFβ, IL10, PGE2 [68]. These immunosuppressive factors also enhance the expression of anti-apoptotic molecules such as BCLxL, cFLIP, MCL1 [69,70]. Galectins are glycan binding proteins that specifically bind to the N-acetyl-lactose amine which are attached to the cell surface by N-linked or O-linked glycans [71]. Galectin-1 expression positively correlates with aggressiveness and metastatic stages of cancer and is predominantly expressed by tumor cells and tumor associated stroma [71]. Galectins-1 induces FAS-L dependent and independent T cell apoptosis, blocking proximal TCR signaling and inhibits the secretion of Th1 type cytokine such as IFNγ, IL2, TNFα [72]. Galectin-1 is one of the major immune suppressive factors that contribute to tumor immune evasion. Galectin-3 restricts lateral movement of TCR complex and thus restrains TCR mediated signaling [72]. In addition several other immunosuppressive factors including Reactive Oxygen Species (ROS), Nitric Oxide (NO), mucins, increased levels of lactate, extracellular adenosine also contribute to immune suppression by targeting immune cells [73].
Dendritic cells
Dendritic Cells (DCs) are most crucial and potent Antigen Presenting Cells (APCs) that recognize take up, process and present tumor antigens to activate T cell specific immune responses. DC can develop from Common Myeloid Progenitor cells (CMPs) or Common Lymphoid Progenitor cell (CLPs) both of which differentiated through common Hematopoietic Progenitor Cells (HPC) [72]. In human, conventional DCs (cDCs) from myeloid origins are predominant. The plasmacytoid DCs (pDCs) which are very low in numbers mostly arise from CLPs but in some occasion CMPs also produce pDCs [72,74]. Although both the DCS originate from same progenitor, their differentiation is controlled by their markers, reprogramming by different stimuli and their specificity to antigens [75]. Activation of DCs leads to differential gene expression, strong co-stimulatory signals such as CD80/CD86/CD40 and release of effector cytokine that stimulate T cell activation. In different human cancers like prostate, breast, and malignant glioma it was found that there is successive loss of pDCs and cDCs whereas coexisting accumulation of iDCs that have reduced antigen processing and presentation capabilities and are unable to elicit IFNγ mediated immune response [72]. Decreased number of functional DCs and increased amount of nonfunctional iDCs causes serious obstacles that lead to tumor progression [76]. Accumulation of iDCs in tumor milieu promotes negative co-stimulatory signal that induces T cell tolerance and energy [77]. It was noticed that in comparison to cDCs, in many cancers such as melanoma and ovarian cancer, pDCs are mostly predominant which suppress T cell activities. Down-regulation of TLR9 and reduced IFNα secretion by pDCs was observed in tumor microenvironment that also contributes tumor antigens to escape from immune surveillance [78]. VEGF, IL10, PGE2, TGFβ are not the only immunesuppresive factors that contributes to impairment of DCs. Recent studies confirms that hypoxia, extracellular adenosine and accumulation of lactate in tumor site also made cDCs and pDCs nonfunctional. Hypoxia Inducible Factor-α (HIF1α) in hypoxic tumor condition induces adenosine receptor A2B in DCs promoting them to stimulate Th2 cells rather than Th1 that’s leads to type-2 cytokine bias [79]. Differentiated DCs in tumorigenic condition and in the presence of adenosine lose their allostimulating activities and produce large amount of IL10, VEGF, IL6, TGFβ, COX2 and IDO [80]. In prostate tumors pDCs modulate intratumorogenic CD8+ T cell function by secreting ARG1 and IDO [81]. IDO producing DCs also induce the suppressive activities of CD4+CD25+FOXP3+ Treg cells that also have a major role in tumor immune evasion [82].
Macrophages
Macrophages are terminally differentiated myeloid cells closely linked to DCs. Immature monocytes are released from the bone marrow and circulate in the blood. They are recruited by chemokines into the tissue and undergo differentiation into macrophages [83]. Tissue macrophages display enormous functional and phenotypic plasticity in response to changing micro-environmental stimuli including cancer [84]. Depending on the diversity displayed by macrophages in terms of receptor expression, cytokine production and functions, it can be classified into two types: Type-1 Macrophages (M1) which are capable of producing large amounts of pro-Inflammatory cytokines (IFNγ, IL12), expressing high levels of MHC molecules, releasing cytotoxic ROS/RNS (reactive oxygen/nitrogen species) and are tumoricidal [85] and Type-2 macrophages which are activated by IL4, IL10, IL13 and glucocorticoid hormones and also secrete high levels of IL10 and very low amount of IL12 that favors tumor progression [86]. The macrophages present in neoplastic tissues are referred to as Tumor-Associated Macrophages (TAMs) and mainly belong to the M2 population [87]. In tumor microenvironment, T cell activation and dysfunctional innate immune responses are also induced when TAM eliminates M1 macrophages from the regime of tumor microenvironment. In the presence of TAM, M1 cannot produce IL12 therefore NK cell, Th1 cell and CTL mediated immune response against tumor is completely abrogated [88]. M2 macrophages secrete profound amount of IL10 that drive Th2 cell development. Th2 cells do not support the development of CTLs and IL4 released from Th2 further induces TAM development [89]. In addition IL10 is required for maintaining Treg cell activities that leads to tumor progression [90]. TAM also released CCL22 which causes Treg trafficking into tumor-site. TAM secretes PGE2, TGFβ and expresses PDL1 that cause immunosuppression and T cell apoptosis [91]. Seven different subsets of TAM have been identified in mouse lung adenocarcinoma and breast cancer based on their receptors which includes LY6C, MHC-II, CX3CR1, CCR2 and CD62L etc. they have different half-lives as well as relative frequencies in tumor progression [92]. MHC-II negative or low TAMs may also induce expression of the angiopoietin receptor TIE2 and they localize to hypoxic sites in tumor. T cells play a major role in regulation of macrophages during tumorigenesis. In mouse mammary adenocarcinoma Th2 cells are predominant that release IL4 and induce TAM of M2 type. This M2-TAMs secret Epidermal Growth Factors (EGFs) which are involved in tumor cell invasion and metastasis.CD4+CD25+CD127lowFOXP3+Treg cells secreting cytokines IL10, IL4, IL13 induces monocytes to differentiate into M2 macrophages by hindering their response to Lipopolysaccharide (LPS). M2 macrophages also induce the expression of CD206 and CD163 that cause polarization of M1 macrophages. In addition to Treg, NKT cells and B cells encourage M2 macrophage generation that produce increase levels of IL10. Tumor cells also contribute to generate M2 macrophages with intense levels of IL10, CCL22, CCL5, Matrix Metalloproteinase-7 (MMP7), MMP9 etc [72,93].
Myeloid-derived (MDSC)
Myeloid-Derived Suppressor Cells (MDSCs) are a heterogeneous population of myeloid cells composed of immature macrophages, granulocytes, dendritic cells and other myeloid cells. In normal condition, immature myeloid cells are generated in bone marrow and differentiate into mature myeloid cells. In cancer, normal pathway of myeloid cell differentiation is hindered which leads to terminal differentiation of mature macrophages, dendritic cells and granulocytes and this generates pathological MDSCs. In human, MDSCs are characterized by expression of CD33, lack expression of HLA-DR and markers of mature lymphoid and myeloid cells. They are hematopoietic progenitors which can differentiate not only into granulocytes, monocytes but also to endothelial cells and osteoclasts [94-99]. Tumor-associated stromal cells form a niche which secrete growth factors such as GMCSF, GCSF, MCSF, Stem Cell Factor (SCF; also known as KIT ligand), VEGF and IL3 to induce myelopoiesis and chemokines such as CCL2, CCL12, CXC-chemokine Ligand-5 (CXCL5), prokineticin-2, S100A8 and S100A9 to mobilize and marginate MDSCs to tumor stroma [100,101,10]. Tumor-derived soluble factors that are pro-inflammatory (like IL1β, IL6, S100A8 and S100A9), as well as cytokines released by activated T cells (i.e. IFNγ, IL4, IL10 and IL13) give birth to MDSCs which initiates various immunosuppressive activities [102,103]. Tumor-derived soluble factors regulate myeloid lineage by expression of transcription factors such as: (a) STAT3 plays an important role in survival, proliferation and differentiation of MDSCs in following manner: (i) up-regulation of BCLxL, survivin, MYC and cyclin D1 [104], (ii) expression of various calcium binding pro-inflammatory protein like S100A8 and S100A9 [105], (iii) it promotes expression of p47phox (also known as NCF1) and p91phox (also known as CYBB) which secrete reactive oxygen species and make MDSCs more suppressive [106], (iv) downregulates protein kinase Cβ isoform-II (PKCβII) which is required for DC differentiation and maturation, up-regulates C/EBPβ [107] (b) STAT1 controls subsets of myeloid cells through its effects on iNOS expression and is crucial for immune suppression by MDSCs [108], (c) STAT6 activates Jumonji Domain containing protein 3 (JMJD3) which escalates expression of ARG1, YM1 and FIZZI1 and finally results to M2 polarization [109]. Additionally NFKβ, COX2 and PGE2 enhances MDSCs generation, accumulation and sterns their suppressive activity [110,111].
NK and NKT cells
NK cells are subsets of innate lymphoid cells that express transcription factors E4BP4 (E4-promoter binding protein-4) and induces apoptosis of tumor cells by secreting IFNγ, perforin, granzyme, or FAS-FASL, TRAIL mediated interaction [112]. NK cells contain inhibitory receptors that interact with MHC-I of selfcells. Tumor cells lacking MHC-I induce hypo-responsiveness of NK cells and promote apoptosis [113]. NK cells also express NKG2D that interact with its ligand MICA and MICB (MHC-I polypeptide-related sequence A/B) present on tumor cells and induce effector responses for their clearance. NK cells contain Fc receptors (CD16) that can bind with antibody coated tumor cells and induce antibody-dependent apoptosis [113,114]. In tumor milieu release of immunosuppressive factors like TGFβ, IDO, PGE2 restricts NK cell activation and contribute NK cell tolerance. NKT cells possess both NK cell and T cell characteristics and restrict tumor cell proliferation [115]. NKT cells express αβ-TCR variant and NK1.1 receptor. NKT type I and II are CD1d restricted response only CD1d expressing tumor cells [116]. Type-I NKT cells express invariant TCRα chain – Vα14 receptor (iNKT cells) and are stimulated by specific glycolipid ligand α-galactosylceramide and release increase levels of IFNγ, perforin, granzyme, and induce FAS-FASL or TRAIL mediated apoptosis of tumor cells. Whereas type-II NKT cells express heterogeneous non- Vα14 receptors and secrete TGFβ, IL13 and activate IL4R-STAT6 signaling that leading to suppression of CTL activities [115,116].
T-regulatory cells
Increased levels of Treg cells were found in different types of cancer such as breast, lung, liver, colorectal as well as melanoma [117]. Treg cells bring tolerance in tumor microenvironment by inhibiting dendritic cell activation, promoting M2-type macrophage induction and induction of T cell apoptosis that triggers immune evasion. Both thymus-derived natural Treg (nTreg) and tumor-induced Treg (iTreg) contributes to tolerance and immunosuppressive activities in tumorogenic condition [118]. FOXP3, the master transcription factor and its associated protein networks play indispensable role for Treg development and function. FOXP3 can act both as a transcriptional activator and repressor. FOXP3 in association with NFATc2 activates CTLA4, CD25, and GITR that promotes suppressive function whereas the same association down-regulates the expression of IL2 and such deprivation of IL2 promotes T-effector cell death [119,120]. In a recent study it has been found that FOXP3 acts as a co-transcription factor with STAT3 that up-regulates IL10 transcription in tumor induced Treg cells [121,122]. Plasmacytoid and myeloid DCs, TAMs and tumor cells release increased levels of TGFβ that convert activated CD4+CD25+ T cells into FOXP3+ Treg cells. Apart from CD25, Treg cells express different context-dependent receptors such as CTLA4, GITR, and PD1 which contribute to T cell cycle arrest and generation of immature APCs such as dendritic cells that are unable to induce effector immune responses against cancer [123]. In tumor milieu tumor cells and TAMs secrete chemokines CCL22 that cause trafficking of CCR4+ Treg cells at tumor site. After trafficking CCL22 can interact with its receptor CCR4 and expansion of Treg occur at tumor site. Treg cells also produce intense levels of Immunoregulatory cytokines IL10 and TGFβ that promote tumor immune evasion by hindering the function of APCs such as DC and T-effector cells [123,124]. Treg cells express CTLA4 that interact with CD80 and CD86 ligand on dendritic cells and constrains dendritic cell function. CTLA4+ Treg cells also secrete IDO that catalyzes tryptophan breakdown and provides decreased co stimulatory signal to DCs. LAG3 expressed in Tregs interact with MHC-II of DCs and limits DC maturation and constrains its effector function as APC. Treg induces CD39 and CD73 that hydrolyze ATP into AMP and adenosine; both of them limits CD80 and CD86 costimulatory signals of DCs and makes them nonfunctional [125]. Nrp1 (neuropilin-1) secreted from FOXP3+ Treg cells interact with immature DCs and alter their functional activities. Treg cells also release granzyme A and B that induce apoptosis of CTLs, T-effector cells, DCs in perforin-dependent and FAS/FASL-independent manner. As IL2 is consumed by Treg cells through its receptor IL2Rα/CD25 that cause IL2 deprivation and induces BIM1-mediated apoptosis of nearby effector cells [125-127] (Figure 4).
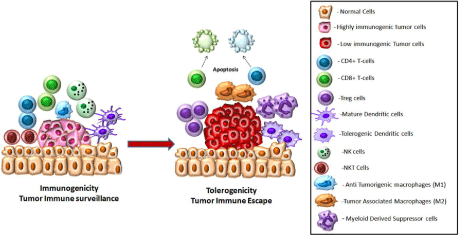
Figure 4: Modulation of different immune cells in the tumor microenvironment.
There is a constant tug of war between the developing tumor and the immune system. Some tumor cells are highly immunogenic and elicit a proper immune response. In such cases effector cells like CD4+and CD8+ T-cells, NK and NK T-cells, anti-tumorigenic macrophages, mature dendritic cells are present in the tumor microenvironment and they secreate cytokines like IFNγ, TNFα, perforin, granzyme. This creates an immunogenic atmosphere and may finally lead to tumor regression. However some tumor cells might be converted into a low immunogenic type which can avoid immune recognition. They recruit tolerogenic cells like Treg, tolerogenic dendritic cells, tumor associated macrophages, myeloid derived dendritic cells. These cells alongwith associated cyokines like IL10, TGFβ, PGE2, Gangliosides, IDO, Galectins create an immunosuppressive environment that promote tumor growth.
Tumor immune escape and inflammation
Tumor cells induces some death receptor ligand such as FASL, that interact with its receptor such as FAS on T cells and triggers cascade of intracellular signaling that cause T cell apoptosis [72,128]. In contrast, expression of FASL in activated T cell and CTLs maintains T cell homeostasis and cytotoxic T cell activities. In some studies it was shown that FASL induces pro-inflammatory and antitumorogenic effects in vivo. Delivery of FASL gene restricts tumor growth instead of tumor immune escape due to the infiltration of inflammatory neutrophils at tumor site [2, 129]. It has been suggested that immune privilege in tumors depends on presence of some immunosuppressive factors such as TGFβ that creates tolerogenic microenvironments to prevent pro-inflammatory FASL and eliminating immune cells that favors tumor growth. Increase levels of FASL induce neutrophil infiltration whereas physiological levels restrict anti-tumor response and contribute tumor immune escape [129]. Tumor also counterattack immune cells by releasing FASL containing micro vesicles during tumor progression and induces apoptosis of FAS-sensitive lymphoid cells [130]. In some cases tumor cells ingest T-effector cells by a process called tumor cannibalism [131]. In addition to FASL some other inhibitory ligand such as TRAIL, RANTES have also been involved in tumor induced immune cell death [132]. Certain tumors induce RCAS1 ligand that facilitates T cell cycle arrest and apoptosis [133]. Gangliosides and CD70-CD27 interaction in some tumors also promote T cell apoptosis [134].
Conclusion
Recent strategies for cancer immunotherapy mainly depend on chemotherapy and vaccination to induce CTL response, introduction of antibodies against immunosuppressive factors and adoptive transfer of T-effector cells to induce tumor regression through tumorimmunogenicity. Although considerable success has been achieved through in vitro studies or preclinical trials but clinical studies have not produced significant results. Tumor microenvironment is composed of different altered immune cells, tumor cells and immunosuppressive factors that create serious obstacles in successful immunotherapy to combat cancer. Many queries regarding immune evasion have still not been answered. Investigation on trafficking of Treg cells, NKT cells, DCs in tumor surroundings or distant sites may provide hopes for successful immunotherapy in future. In addition blockade of negative inhibitory signals together with conventional therapy also needing further exploration. Recently several combined immune strategies such as blockade of CTLA4 with GM-CSF secreting vaccine, or chemotherapy with IDO blockade has been developed which provides some sort of effective immune response against cancer. Removal of inhibitory signals and reshaping the immune cells so that they target tumor cells by combined immunotherapy will be successful to overcome tumorigenic tolerance in future scenario.
Acknowledgement
This work was supported by grants from the Department of Biotechnology and Council for Scientific and Industrial Research and Department of Science and Technology, Government of India.
References
- Smyth MJ, Godfrey DI, Trapani JA. A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol. 2001; 2: 293-299.
- Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002; 3: 999-1005.
- Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol. 2003; 21: 807-839.
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002; 3: 991-998.
- Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004; 22: 329-360.
- Rosenberg SA. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity. 1999; 10: 281-287.
- Spiotto MT, Yu P, Rowley DA, Nishimura MI, Meredith SC, Gajewski TF, et al. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity. 2002; 17: 737-747.
- Ochsenbein AF, Klenerman P, Karrer U, Ludewig B, Pericin M, Hengartner H, et al. Immune surveillance against a solid tumor fails because of immunological ignorance. Proc Natl Acad Sci USA. 1999; 96: 2233-2238.
- Palmowski M, Salio M, Dunbar RP, Cerundolo V. The use of HLA class I tetramers to design a vaccination strategy for melanoma patients. Immunol Rev. 2002; 188: 155-163.
- Degli-Esposti MA, Smyth MJ. Close encounters of different kinds: dendritic cells and NK cells take centre stage. Nat Rev Immunol. 2005; 5: 112-124.
- Harizi H. Reciprocal crosstalk between dendritic cells and natural killer cells under the effects of PGE2 in immunity and immunopathology. Cell Mol Immunol. 2013; 10: 213-221.
- Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. 2013; 25: 214-221.
- Sinha P, Ghosh AK, Das T, Sa G, Ray PK. Protein A of Staphylococcus aureus evokes Th1 type response in mice. Immunol Lett. 1999; 67: 157-165.
- Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. Int J Biol Sci. 2011; 7: 651-658.
- Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune microenvironments in solid tumors: new targets for therapy. Genes Dev. 2011; 25: 2559-2572.
- Hadrup S, Donia M, Thor Straten P. Effector CD4 and CD8 T cells and their role in the tumor microenvironment. Cancer Microenviron. 2013; 6: 123-133.
- Schmid DA, Irving MB, Posevitz V, Hebeisen M, Posevitz-Fejfar A, Sarria JC, et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J Immunol. 2010; 184: 4936-4946.
- Cole DK, Pumphrey NJ, Boulter JM, Sami M, Bell JI, Gostick E, et al. Human TCR-binding affinity is governed by MHC class restriction. J Immunol. 2007; 178: 5727-5734.
- Colonna M, Cella M. Crosspresentation: plasmacytoid dendritic cells are in the business. Immunity. 2007; 27: 419-421.
- Baitsch L, Fuertes-Marraco SA, Legat A, Meyer C, Speiser DE. The three main stumbling blocks for anticancer T cells. Trends Immunol. 2012; 33: 364-372.
- Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. 2013; 25: 214-221.
- Wherry EJ. T cell exhaustion. Nat Immunol. 2011; 12: 492-499.
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006; 439: 682-687.
- Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003; 22: 4212-4222.
- Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009; 11: 973-979.
- Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol. 2011; 11: 289-295.
- Pawelec G, Solana R. Immunoageing - the cause or effect of morbidity. Trends Immunol. 2001; 22: 348-349.
- Fearon DT, Manders P, Wagner SD. Arrested differentiation, the selfrenewing memory lymphocyte, and vaccination. Science. 2001; 293: 248- 250.
- Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat Med. 2005; 11: 1299-1305.
- Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol. 2000; 74: 181-273.
- Töpfer K, Kempe S, Müller N, Schmitz M, Bachmann M, Cartellieri M, et al. Tumor evasion from T cell surveillance. J Biomed Biotechnol. 2011; 2011: 918471.
- Rivoltini L, Carrabba M, Huber V, Castelli C, Novellino L, Dalerba P, et al. Immunity to cancer: attack and escape in T lymphocyte-tumor cell interaction. Immunol Rev. 2002; 188: 97-113.
- Wang Z, Cao Y, Albino AP, Zeff RA, Houghton A, Ferrone S. Lack of HLA class I antigen expression by melanoma cells SK-MEL-33 caused by a reading frameshift in beta 2-microglobulin messenger RNA. J Clin Invest. 1993; 91: 684-692.
- Mizoguchi H, O’Shea JJ, Longo DL, Loeffler CM, McVicar DW, Ochoa AC. Alterations in signal transduction molecules in T lymphocytes from tumorbearing mice. Science. 1992; 258: 1795-1798.
- Koneru M, Schaer D, Monu N, Ayala A, Frey AB. Defective proximal TCR signaling inhibits CD8+ tumor-infiltrating lymphocyte lytic function. J Immunol. 2005; 174: 1830-1840.
- Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009; 229: 12-26.
- Hathcock KS, Laszlo G, Dickler HB, Bradshaw J, Linsley P, Hodes RJ. Identification of an alternative CTLA-4 ligand costimulatory for T cell activation. Science. 1993; 262: 905-907.
- Linsley PS, Clark EA, Ledbetter JA. Pillars article: T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/ BB-1. 1990. Proc. Natl. Acad. Sci. USA 87: 5031-5035. J Immunol. 2009; 182: 2559-2563.
- Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994; 1: 793-801.
- Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 2002; 16: 23-35.
- Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cellextrinsic function of CTLA-4. Science. 2011; 332: 600-603.
- Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008; 322: 271-275.
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000; 192: 1027-1034.
- Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999; 5: 1365-1369.
- Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001; 193: 839-846.
- Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009; 206: 3015-3029.
- Sfanos KS, Bruno TC, Meeker AK, De Marzo AM, Isaacs WB, Drake CG. Human prostate-infiltrating CD8+ T lymphocytes are oligoclonal and PD-1+. Prostate. 2009; 69: 1694-1703.
- Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007; 13: 84-88.
- Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci USA. 2008; 105: 20852-20857.
- Kim J, Myers AC, Chen L, Pardoll DM, Truong-Tran QA, Lane AP, et al. Constitutive and inducible expression of b7 family of ligands by human airway epithelial cells. Am J Respir Cell Mol Biol. 2005; 33: 280-289.
- Lee SK, Seo SH, Kim BS, Kim CD, Lee JH, Kang JS, et al. IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J Dermatol Sci. 2005; 40: 95-103.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012; 12: 252-264.
- He C, Qiao H, Jiang H, Sun X. The inhibitory role of b7-h4 in antitumor immunity: association with cancer progression and survival. Clin Dev Immunol. 2011; 695834.
- Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009; 10: 29-37.
- Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005; 6: 1245-1252.
- Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. 2005; 6: 90-98.
- Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006; 24: 99-146.
- Thomas DA, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005; 8: 369- 380.
- Kim R, Emi M, Tanabe K, Uchida Y, Toge T. The role of Fas ligand and transforming growth factor h in tumor progression: molecular mechanisms of immune privilege via Fas-mediated apoptosis and potential targets for cancer therapy. Cancer. 2004; 100: 2281–2291.
- Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001; 166: 678- 689.
- Harizi H, Juzan M, Grosset C, Rashedi M, Gualde N. Dendritic cells issued in vitro from bone marrow produce PGE(2) that contributes to the immunomodulation induced by antigen-presenting cells. Cell Immunol. 2001; 209: 19-28.
- Kurte M, Lopez M, Aguirre A, Escobar A, Aguillon JC, Charo J, et al. A synthetic peptide homologous to functional domain of human IL-10 downregulates expression of MHC class I and transporter associated with antigen processing 1/2 in human melanoma cells. J Immunol. 2004; 173: 1731–1737.
- Mellor AL, Munn DH. Immunology at the maternal-fetal interface: lessons for T cell tolerance and suppression. Annu Rev Immunol. 2000; 18: 367-391.
- Hossain DM, Mohanty S, Ray P, Das T, Sa G. Tumor gangliosides and T cells: a deadly encounter. Front Biosci (Schol Ed). 2012; 4: 502-519.
- Birklé S, Zeng G, Gao L, Yu RK, Aubry J. Role of tumor-associated gangliosides in cancer progression. Biochimie. 2003; 85: 455-463.
- Zhao J, Furukawa K, Fukumoto S, Okada M, Furugen R, Miyazaki H, et al. Attenuation of interleukin 2 signal in the spleen cells of complex gangliosidelacking mice. J Biol Chem. 1999; 274: 13744-13747.
- Kim R, Emi M, Tanabe K, Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res. 2006; 66: 5527-5536.
- Kim R, Emi M, Tanabe K. Cancer cell immune escape and tumor progression by exploitation of anti-inflammatory and pro-inflammatory responses. Cancer Biol Ther. 2005; 4: 924-933.
- Espaa L, Fernández Y, Rubio N, Torregrosa A, Blanco J, Sierra A. Overexpression of Bcl-xL in human breast cancer cells enhances organselective lymph node metastasis. Breast Cancer Res Treat. 2004; 87: 33-44.
- Jung YJ, Kim JY, Park JH. TGF-beta1 inhibits Fas-mediated apoptosis by regulating surface Fas and cFLIPL expression in human leukaemia/ lymphoma cells. Int J Mol Med. 2004; 13: 99-104.
- Camby I, Le Mercier M, Lefranc F, Kiss R. Galectin-1: a small protein with major functions. Glycobiology. 2006; 16: 137R-157R.
- Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007; 25: 267-296.
- Koido S, Homma S, Takahara A, Namiki Y, Tsukinaga S, Mitobe J, et al. Current immunotherapeutic approaches in pancreatic cancer. Clin Dev Immunol. 2011; 267539.
- Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002; 2: 151-161.
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012; 12: 253-268.
- Enk AH, Jonuleit H, Saloga J, Knop J. Dendritic cells as mediators of tumorinduced tolerance in metastatic melanoma. Int J Cancer. 1997; 73: 309-316.
- Nestle FO, Burg G, Fah J, Wrone-Smith T, Nickoloff BJ. Human sun light induced basal-cell-carcinoma-associated dendritic cells are deficient in T cell costimulatory molecules and are impaired as antigen-presenting cells. Am J Pathol. 1997; 150: 641–651.
- Hartmann E, Wollenberg B, Rothenfusser S, Wagner M, Wellisch D, Mack B, et al. Identification and functional analysis of tumor-infiltrating plasmacytoid dendritic cells in head and neck cancer. Cancer Res. 2003; 63: 6478-6487.
- Yang M, Ma C, Liu S, Shao Q, Gao W, Song B, et al. HIF-dependent induction of adenosine receptor A2b skews human dendritic cells to a Th2- stimulating phenotype under hypoxia. Immunol Cell Biol. 2010; 88: 165-171.
- Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood. 2008; 112: 1822-1831.
- Watkins SK, Zhu Z, Riboldi E, Shafer-Weaver KA, Stagliano KE, Sklavos MM, et al. FOXO3 programs tumor-associated DCs to become tolerogenic in human and murine prostate cancer. J Clin Invest. 2011; 121: 1361-1372.
- Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, et al. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol. 2009; 183: 2475-2483.
- Hunter M, Wang Y, Eubank T, Baran C, Nana-Sinkam P, Marsh C. Survival of monocytes and macrophages and their role in health and disease. Front Biosci (Landmark Ed). 2009; 14: 4079-4102.
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008; 8: 958-969.
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002; 23: 549-555.
- Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol. 2009; 70: 325-330.
- Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumorassociated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med. 2010; 362: 875-885.
- Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010; 32: 593-604.
- DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009; 16: 91-102.
- Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. 2009; 10: 1178-1184.
- Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009; 206: 1327-1337.
- Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van den Bossche J, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6Chigh monocytes. Cancer Res. 2010; 70: 5728–5739.
- Hagemann T, Wilson J, Burke F, Kulbe H, Li NF, Plüddemann A, Charles K. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J Immunol. 2006; 176: 5023-5032.
- Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010; 70: 68-77.
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009; 9: 162-174.
- Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001; 61: 4756-4760.
- Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. 2011; 208: 1949-1962.
- Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGFbeta 1. J Immunol. 2009; 182: 240-249.
- Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumorinduced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006; 66: 1123-1131.
- Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007; 450: 825-831.
- Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008; 13: 23-35.
- Huang B, Lei Z, Zhao J, Gong W, Liu J, Chen Z, et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 2007; 252: 86-92.
- Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006; 176: 284-290.
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009; 9: 162-174.
- Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008; 205: 2235- 2249.
- Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloidderived suppressor cells. J Immunol. 2009; 182: 5693-5701.
- Farren MR, Carlson LM, Lee KP. Tumor-mediated inhibition of dendritic cell differentiation is mediated by down regulation of protein kinase C beta II expression. Immunol Res. 2010; 46: 165-176.
- Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol. 2005; 174: 4880-4891.
- Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood. 2009; 114: 3244-3254.
- Martino A, Badell E, Abadie V, Balloy V, Chignard M, Mistou MY, et al. Mycobacterium bovis bacillus Calmette-Guérin vaccination mobilizes innate myeloid-derived suppressor cells restraining in vivoT cell priming via IL 1R dependent nitric oxide production. J Immunol. 2010; 184: 2038–2047.
- Zhang Y, Liu Q, Zhang M, Yu Y, Liu X, Cao X. Fas signal promotes lung cancer growth by recruiting myeloid-derived suppressor cells via cancer cellderived PGE2. J Immunol. 2009; 182: 3801-3808.
- Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012; 12: 239-252.
- Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011; 331: 44–49.
- Han Z, Jing Y, Zhang S, Liu Y, Shi Y, Wei L. The role of immunosuppression of mesenchymal stem cells in tissue repair and tumor growth. Cell Biosci. 2012; 2: 8.
- Kronenberg M, Gapin L. The unconventional lifestyle of NKT cells. Nat Rev Immunol. 2002; 2: 557-568.
- Terabe M, Matsui S, Noben-Trauth N, Chen H, Watson C, Donaldson DD, et al. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat Immunol. 2000; 1: 515-520.
- Darrasse-Jèze G, Podsypanina K. How numbers, nature, and immune status of foxp3(+) regulatory T-cells shape the early immunological events in tumor development. Front Immunol. 2013; 4: 292.
- Banerjee A, Vasanthakumar A, Grigoriadis G. Modulating T regulatory cells in cancer: how close are we? Immunol Cell Biol. 2013; 91: 340-349.
- Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol. 2012; 13: 1010-1019.
- Rudensky AY, Gavin M, Zheng Y. FOXP3 and NFAT: partners in tolerance. Cell. 2006; 126: 253-256.
- Hossain DM, Panda AK, Manna A, Mohanty S, Bhattacharjee P, Bhattacharyya S, et al. FoxP3 acts as a cotranscription factor with STAT3 in tumor-induced regulatory T cells. Immunity. 2013; 39: 1057-1069.
- Bhattacharyya S, Md Sakib Hossain D, Mohanty S, Sankar Sen G, Chattopadhyay S, Banerjee S, et al. Curcumin reverses T cell-mediated adaptive immune dysfunctions in tumor-bearing hosts. Cell Mol Immunol. 2010; 7: 306-315.
- Hossain DM, Panda AK, Chakrabarty S, Bhattacharjee P, Kajal K, Mohanty S, et al. MEK inhibition prevents tumour-shed transforming growth factor-β- induced T-regulatory cell augmentation in tumour milieu. Immunology. 2015; 144: 561-573.
- von Boehmer H, Daniel C. Therapeutic opportunities for manipulating T(Reg) cells in autoimmunity and cancer. Nat Rev Drug Discov. 2013; 12: 51-63.
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008; 133: 775-787.
- Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009; 30: 636-645.
- Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013; 138: 105-115.
- O’Connell J, Houston A, Bennett MW, O’Sullivan GC, Shanahan F. Immuneprivilege or inflammation? Insights into the Fas ligand enigma. Nature Med. 2001; 7: 271–274.
- Chen JJ, Sun Y, Nabel GJ. Regulation of the proinflammatory effects of Fas ligand (CD95L). Science. 1998; 282: 1714-1717.
- Andreola G, Rivoltini L, Castelli C, Huber V, Perego P, Deho P, et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med. 2002; 195: 1303-1316.
- Lugini L, Matarrese P, Tinari A, Lozupone F, Federici C, Iessi E, et al. Cannibalism of live lymphocytes by human metastatic but not primary melanoma cells. Cancer Res. 2006; 66: 3629-3638.
- Giovarelli M, Musiani P, Garotta G, Ebner R, Di Carlo E, Kim Y, et al. A “stealth effect”: adenocarcinoma cells engineered to express TRAIL elude tumor-specific and allogeneic T cell reactions. J Immunol. 1999; 163: 4886- 4893.
- Nakashima M, Sonoda K, Watanabe T. Inhibition of cell growth and induction of apoptotic cell death by the human tumor-associated antigen RCAS1. Nat Med. 1999; 5: 938-942.
- Raval G, Biswas S, Rayman P, Biswas K, Sa G, Ghosh S, et al. TNF-alpha induction of GM2 expression on renal cell carcinomas promotes T cell dysfunction. J Immunol. 2007; 178: 6642-6652.