
Abstract
KBG syndrome is a rare genetic disorder that presents with variable clinical features involving multiple systems and is caused by pathogenic variants in the ANKRD11 gene. The KIAA1109-related syndrome is a rare autosomal recessive inherited disorder caused by pathogenic variants in the KIAA1109 gene and presents with severe developmental delay, brain malformations, and seizures. This case report presents a rare and unique neonatal presentation of KBG syndrome associated with a novel genetic mutation of KIAA1109-related syndrome. The patient’s clinical features, investigations, treatment, and follow-up are discussed, shedding light on the coexistence of two distinct genetic conditions in a single individual.
Introduction
KBG syndrome and KIAA1109-related syndrome are distinct and rare genetic disorders, each presenting unique clinical features and underlying genetic mutations. Both syndromes have significant implications for affected individuals and their families, necessitating early diagnosis and personalized management approaches. Moreover, due to the range of symptoms and variable expression patterns observed in these syndromes, they are often underdiagnosed [3].
KBG syndrome is a sporadic genetic disorder with a broad spectrum of clinical manifestations affecting multiple systems. There have only around 200 reported cases, in the literature; thus, diagnosing early without genetic testing is challenging [3]. The acronym "KBG" stands for the initials of the last names of three original families of the cases reported [1]. Variation in the ANKRD11 gene mainly caused craniofacial anomalies, growth and developmental anomalies, skeletal system anomalies, and nervous system anomalies. Notable common features in affected individuals include enlarged teeth (macrodontia), triangular head shape (brachycephaly), and a prominent nasal tip. Other characteristics include facial features, like eyes spaced far apart from eyebrows with an arch and a wide nasal bridge [4]. They may also present with neurodevelopmental delays and intellectual disability of varying degrees [4]. The primary cause of KBG syndrome is pathogenic variants in the ANKRD11 gene, located on chromosome 16q24.3. These mutations typically occur de novo, where they arise spontaneously and are not inherited from either parent [4].
The KIAA1109-related syndrome, also known as Alkuraya-Kucinskas syndrome, is another rare autosomal recessive genetic disorder characterized by severe developmental delay, brain malformations, and seizures [2]. This syndrome has been recently recognized, and only limited data regarding its prevalence and clinical features is available. However, it’s crucial to be aware of this syndrome in individuals experiencing developmental delays and neurological issues. The KIAA1109-related syndrome is caused by loss of function mutations in the KIAA1109 gene, which is involved in brain development and function [2]. These patients have developmental delay due to brain malformations and present with muscle and joint stiffness throughout the body [2].
Our case report presents the rare neonatal presentation of KBG syndrome associated with an unprecedented genetic mutation of KIAA1109-related syndrome, offering a unique insight into the coexistence of these two rare conditions in one patient.
Case Presentation
We present the case of a male neonate born by spontaneous vaginal delivery at 38+2/7 weeks' gestation with birth weight of 3.1 kg. The mother had a history of ADHD and was on stimulant Focallin (Dexmethylphenidate) before and during pregnancy. Due to concerns about brain anomalies on prenatal MRI like marked hypogenesis of corpus callosum, enlargement of the bilateral medial ganglionic eminences, globally delayed gyral sulcal pattern with irregularity of ependymal surfaces, abnormal brainstem morphology, comprehensive prenatal testing was conducted including amniocentesis and single nucleotide polymorphism microarray analysis. Both of the tests showed normal chromosomes and no significant chromosomal imbalances. At birth, the baby had a weak cry with central cyanosis requiring suction and tactile stimulation and required to be started on CPAP. Apgar scores are 2 at 1 minute and 8 at 5 minutes, portraying initial challenges in adaptation. Prolonged and refractory seizures were noticed on the day of life #1, requiring multiple anti-epileptic drugs such as Phenobarbitone, Keppra, and Fosphenytoin to control.
The neonate also presented with a distinctive array of clinical features, including small anterior fontanelle, up-slanting palpebral fissures, hypertelorism, micrognathia, thin lips, wide philtrum, and low-set ears. Bilateral cataracts were detected during the initial eye examination and bilateral conductive hearing loss was noticed during the initial hearing screen. Head ultrasound demonstrated multiple brain abnormalities including corpus callosum dysgenesis, ventriculomegaly, multiple germinolytic cysts, diffuse abnormal gyral sulcation pattern, abnormal pituitary gland, and features indicative of cerebellar hypoplasia. Post-natal brain MRI w/o contrast showed stable appearance of the brain with severe lissencephaly/agyria and cerebellar dysplasia as shown in Figure 1. MRI of the eyes indicated optic nerve hypoplasia. This severe abnormality of neuronal migration exhibited in this case is not typical of previously reported brain imaging findings in KBG syndrome. This suggests that the patient’s brain malformation may be a manifestation of another genetic abnormality. Additional investigations, including ECHO, polysomnography, and micro-laryngo bronchoscopy provided further insights into cardiac, respiratory, and upper airway anomalies. Though, pre-natal amniocentesis revealed normal chromosomes, genetic tests like exome sequencing revealed a maternally inherited pathogenic variant in the ANKRD11 gene resulting in KBG syndrome and a paternally inherited pathogenic variant in KIAA1109 causing KIAA1109-related syndrome.
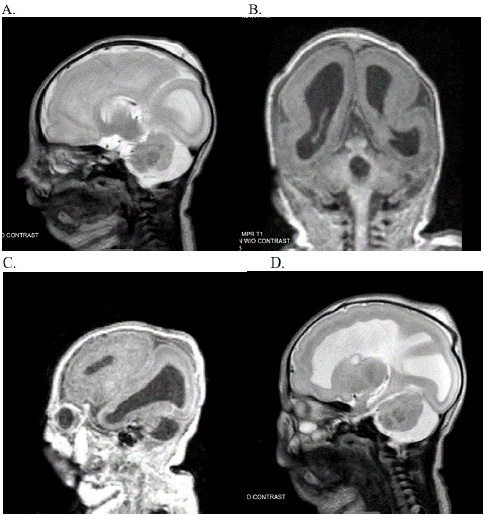
Figure 1: Brain MRI images. (A) severe lissencephaly/agyria and frontal heterotopy in the white matter (B) cerebellar dysplasia Frontal heterotopy in the white matter near the lateral ventricles and in the temporo-basal region. (C-D): Optic nerve hypoplasia.
The neonate required multiple doses of phenobarbitone 22 mg (5.6 mL via G tube every 12 hrs.), levetiracetam 340 mg (3.4 mL via G tube every 12 hrs), and fosphenytoin for seizure management followed by maintenance therapy with lacosamide 42 mg (4.2 mL via G tube every 12 hours), and oxcarbazepine. He received respiratory support with bubble CPAP during admission, gradually transitioning to a High-Flow nasal cannula and room air over one month. Bilateral cataract extraction with anterior vitrectomy was performed. Upon discharge, the neonate was prescribed a regimen of medications, 1% topical atropine, 10 mcg/mL cholecalciferol, 10 mg/mL lacosamide solution, levetiracetam 100 mg/mL solution, 0.5% topical moxifloxacin, 20 mg/ 5 mL phenobarbital elixir, and sulfamethoxazole-trimethoprim 200-40 mg/5 mL suspension. The neonate was also scheduled to follow up with a Pediatrician, Neurologist, Geneticist, and Cardiologist on an outpatient basis.
Discussion
This case highlights the clinical features resulting from the co-existence of two different genetic diseases, KBG syndrome and KIAA1109-related syndrome. This convergence of two distinct genetic conditions emphasizes the complexity of the genetic interactions and their impact on clinical phenotypes.
The early diagnosis of the two rare genetic conditions in the neonate with overlapping complex clinical presentation is relatively new in medical literature. This also raises intriguing questions about the interplay between various genetic pathways contributing to different clinical phenotypes. This observation challenges the traditional one gene, one syndrome paradigm and emphasizes the better understanding of different genetic interactions in shaping clinical phenotypes.
Despite normal findings in prenatal diagnostic tests, post-natal genetic analyses, particularly exome sequencing played a significant role in identifying the genetic etiology of the disease. This highlights the importance of genetic studies in early diagnosis, personalized management, facilitating targeted intervention, leading to a better prognosis.
Enhanced understanding and research into genetic interactions and their impact on disease phenotypes is needed to advance the diagnostic accuracy tailoring targeted therapeutics to individual patients. This case also helps health care providers by raising awareness of the potential overlap of two genetic conditions, where traditional diagnostic methods give inconclusive results.
Conclusion
This case report unveils a compelling instance where a neonatal presentation of KBG syndrome presenting with a novel mutation associated with KIAA1109-related syndrome underscoring the complexity of genetic interactions and their influence on clinical phenotypes. This observation encourages further research into genetic overlaps, interactions, and their contribution to disease expression. Understanding the nuanced interplay between genetic variations will enhance diagnostic accuracy and personalized care, and ultimately contribute to advancements in clinical genetics. This case has also highlighted the importance of genetic study in identifying disease early, helping in better understanding of the disease pathogenesis and prognosis. The long-term outcomes and implications of the identified genetic mutations required continued monitoring and intervention.
References
- Herrmann J, Pallister PD, Tiddy W, Opitz JM. The KBG syndrome- a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig Artic Ser. 1975; 11: 7–18.
- Gueneau L, Fish RJ, Shamseldin HE, Voisin N, Tran Mau-Them F, Preiksaitiene E, et al. KIAA1109 Variants Are Associated with a Severe Disorder of Brain Development and Arthrogryposis. The American Journal of Human Genetics. 2018; 102: 116–32.
- Morel Swols D, Foster J, Tekin M. KBG syndrome. Orphanet J Rare Dis. 2017; 12: 183.
- Sirmaci A, Spiliopoulos M, Brancati F, Powell E, Duman D, Abrams A, et al. Mutations in ANKRD11 Cause KBG Syndrome, Characterized by Intellectual Disability, Skeletal Malformations, and Macrodontia. AmericanJournal of Human Genetics. 2011; 89: 289–94.