
Special Article - Depression Disorders & Treatment
Ann Depress Anxiety. 2016; 3(1): 1076.
Ion Dysregulation in the Pathogenesis of Bipolar Illness
El-Mallakh RS*, Timothy Yff and Yonglin Gao
Department of Psychiatry and Behavioral Sciences, University of Louisville School of Medicine, USA
Corresponding author: El-Mallakh RS, Mood Disorders Research Program, Department of Psychiatry and Behavioral Sciences, University of Louisville School of Medicine, 401 E. Chestnut Street, Suite 610, University of Louisville HealthCare OutPatient Center, Louisville, Kentucky, 40202, USA
Received: March 23, 2016; Accepted: May 23, 2016; Published: May 27, 2016
Abstract
Background: Bipolar disorder is a severe, enigmatic condition that continues to be poorly understood and difficult to treat. True advances in the improvement of the prognosis of this condition will quickly follow insight into its pathogenesis. Ion dysregulatory abnormalities have remained among the most reproducible pathophysiologic alterations in this disease.
Methods: A directed review of studies examining the pathophysiology of bipolar illness was performed.
Results: Several lines of evidence support a central role of ion dysregulation in the pathogenesis of this disorder. Over 75% of all genes associated with bipolar illness are genes that control ion regulation. Measures of intracellular sodium and calcium reveal consistent abnormalities. Endogenous regulators of ion pumps appear to be dysregulated in bipolar patients. All effective agents share common mechanisms of reducing neuronal sodium influx and cellular excitability. And modeling these abnormalities in animals and in vitro produces manic-like, depressive-like behaviors, and cycling.
Conclusion: A model is presented by which ion dysregulation can produce most of the characteristics of bipolar disorder.
Keywords: Bipolar disorder; Pathophysiology; Pathogenesis; Ions; Sodium; Calcium; Potassium
Introduction
Bipolar illness is a severe psychiatric condition that manifests as episodes of mania and depression, interspersed within a baseline that is initially normal but declines as a function of the duration of time spent ill [1]. Effective treatments are available, but effectiveness is suboptimal, and social and occupational dysfunction is a common outcome [1]. The major deterrent to developing more effective treatments is inadequate understanding of the pathophysiology and pathogenesis of the illness. Pathophysiology describes biochemical changes that occur during the ill phases of the disorder. Pathogenesis alludes to the actual cause of the disorder – the ‘primary fault’ that results in the cascade of brain events that produce mania, depression, and other features of the disorder. While research into both of these arenas is limited, synthesis of the available research is nearly absent. This review will focus on ion flux dysregulation as one of the most promising aspects to understanding the disorder.
Abnormalities in the transport and intracellular concentrations of several ions have been repeatedly reported in bipolar disorder. Specifically, nearly 75% of the susceptibility loci that have been linked to bipolar illness include genes that are involved in ion regulation [2]. Measures of intracellular sodium and calcium reveal consistent abnormalities [3,4]. Endogenous regulators of ion pumps appear to be dysregulated in bipolar patients [5]. All effective agents share common mechanisms of reducing neuronal sodium influx and cellular excitability [6,7]. And modeling these abnormalities in animals and in vitro produces manic-like, depressive-like behaviors, and cycling [8-10]. This review will examine the literature regarding ion dysregulation in bipolar illness.
Methods
This was a directed review, which means that specific topics within the area of ions and bipolar illness were specifically reviewed. Literature regarding ions and genes/genetics, intracellular ion perturbations, endogenous regulators of ion channels and pumps, role of ion dysregulation in cellular/neuronal function and imaging, mechanisms of action of effective mood stabilizing agents, and animal models of bipolar illness was searched and reviewed. Two databases, PubMed and Google Scholar, were used.
Results and Discussion
Early studies
Initial interest in the role of ion dysregulation in bipolar illness began shortly after the demonstration that lithium, a monovalent cation, was effective in the treatment of bipolar illness [11]. The 1950s was a time of significant advances in the understanding of neuronal function – how resting and threshold potentials are maintained, and what causes neurons to fire [12] as well as the discovery of the sodium pump and ion channels [13]. It was thus a natural connection to study ion dysregulation in bipolar patients. Those early studies focused on peripheral red and white blood cells. Experiments were performed in “metabolic units”, where research subjects were maintained for weeks in environments in which the total intake of all important cations was carefully controlled. In these experiments it was found that intraerythrocyte sodium concentrations were elevated in manic patients [14,15]. Studies utilizing whole body distribution of radioactive sodium (24Na), potassium (40K), and bromine (82Br) to calculate concentrations in extra vascular compartments, determined that intracellular sodium is increased throughout the body of ill bipolar patients but not euthymic patients [3,16]. The increases of “residual sodium” (which included intracellular sodium and a small fraction of bone sodium) in mania were 400% that of control, and in depression some 200% of control [3]. These values normalized with treatment.
These studies led to a large number of subsequent studies that looked at the activity of the sodium- and potassium-activated adenosine triphosphatase pump (the sodium pump or Na, K-AT Pase) in erythrocyte membranes of bipolar patients [17-19]. A metaanalysis of these studies found that sodium pump activity was indeed reduced in manic and depressed patients compared to both euthymic bipolar subjects and non-bipolar controls [20]. The reduction of pump activity compared to euthymic bipolar subjects was greater in depressed patients (effect size -0.62, confidence interval -1.01 to -0.23, p = 0.002) than in manic patients (effect size -0.42, confidence interval -0.69 to -0.15, p = 0.002) [20]. These findings, among others, were the basis for the introduction of the sodium pump hypothesis for bipolar illness (Figure 1) [21]. This hypothesis argued that a slight reduction in sodium pump activity resulted in manic symptoms, whereas a greater reduction could result in depressive symptoms and catatonia (Figure 1).
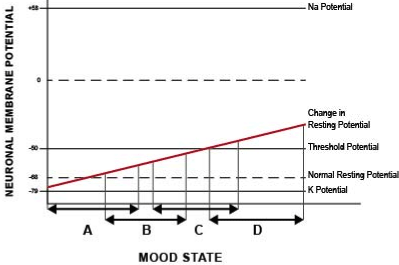
Figure 1: The sodium pump hypothesis purports that as sodium pump activity
decreases and intracellular sodium increases, one of the changes that occurs
is that the resting potential moves closes to the threshold potential. Thus, the
two poles of bipolar illness are actually related to a monopolar pathologic
process in which resting potential departs from the normal range (A).
Slightly depolarized neurons are easier to stimulate and are less regulated,
corresponding to manic symptoms (B). When a fraction of neurons enters
into depolarization block, depressive symptoms appear (C). When a large
number of neurons enter into depolarization block, the patient appears to be
catatonic (D) [21].
Genetic and ion channel studies
Genetic studies have been frustratingly vague. Many associated genetic anomalies have not been reproducible. Nonetheless, nearly 75% of these genes that have been identified are involved in ion transport and regulation [2]. The most significant of these have been genes directly involved in ion regulation. These include CACNA1C (calcium channel, voltage-dependent, L type, alpha 1C subunit) [22], ANK3 (ankyrin 3 or node of Ranvierankyrin G) [22-25], and KCNQ2 (Kv7.2, voltage gated KQT-like subfamily Q, member 2, potassium channel) [26-28]. CACNA1C is a calcium pump protein that is involved in the regulation of intracellular calcium concentration. Variants that have been associated with bipolar illness are associated with increased basal intracellular calcium in bipolar patients [29]. Ankyrin is a cytoskeletal protein that is essential for binding of ion pumps and ion channels [27]. KCNQ2 has binding motifs for the ankyrinprotein; binding to this cytoskeletal protein allows the potassium channels to be localized in the proper area of the membrane and to function appropriately [30]. Along with KCNQ3 (Kv7.3), KCNQ2 interacts with the ankyrin at the first axonal segment to inhibit repetitive firing to prevent neuronal hyperactivity [27,31]. Reducing activity of KCNQ2 in transgenic mice induces hyperactivity, cognitive decline, and neuronal hyper excitability [32]. Lithium may indirectly affect the protein products of KCNQ2 and KCNQ3 by inhibiting their phosphorylation via inhibition of GSKβ (which phosphorylates Kv7.2 and Kv7.3) [26].
Endogenous sodium pump regulators
Early measures of increased intracellular sodium and reduced sodium pump activity were generally performed on circulating red blood cells. Two factors, the first being that erythrocytes do not possess a nucleus, and the second being that these changes are clearly mood-state (not trait) related, suggest that some circulating factor is responsible for these changes. The Na, K-ATPase is known to be the physiologic target for cardiac glycosides, digoxin and ouabain. More recently, it has been discovered that cardenolides with chemical structures very similar to the plant-derived digoxin and ouabain [33,34] are made endogenously in the adrenal and central nervous system of mammals [35,36]. These endogenous cardenolides function in the body the same as their plant-derived look-alikes. Specifically, they have a biphasic curve where low, physiologic concentrations stimulate the sodium pump, while higher, pharmacologic concentrations inhibit the pump [37,38]. Consequently, if the reduced activity of erythrocyte Na, K-ATPase in mania and depression has anything to do with circulating endogenous cardenolides, one would expect reduced circulating levels of these cardenolides.
Euthymic type I bipolar subjects have reduced levels of the Ouabain-Life Factor (OLF) [39]. But more importantly bipolar patients are unable to increase levels of ouabain-like factors at times that normal control subjects have increased levels of OLF. Specifically, exercise to exhaustion is known to increase levels of endogenous cardenolides [40], however, when euthymic bipolar patients are instructed to exercise to exhaustion, they are unable to exercise as extensively as non-bipolar controls [41], and they produce lower levels of OLF [39]. Similarly, it has been found that endogenous cardenolide levels normally have a seasonal pattern in which levels are low in the winter, but significantly higher in the spring, summer, and autumn [42]. However, bipolar patients have lower, winterlike levels throughout the entire seasonal cycle [42]. Inability to regulate production of endogenous cardenolides increases the risk for bipolar patients to have lower levels of these cardenolides when there is a physiologic need to have higher levels. For example, in normal pregnancy there is an increase in the endogenous OLF and a corresponding drop in blood pressure (because at physiologic concentrations OLF increases sodium pump activity) [43,44]. This higher concentration of OLF drops rapidly within 3 -5 days of delivery, but remain higher than baseline [44]. OLF in pregnant women with bipolar has not been measured, but would be expected to be lower during pregnancy – possibly playing a role in making such pregnancies higher risk – and to drop faster than occurs in non-bipolar subjects – possibly increasing the risk for post-partum depression or psychosis.
Clinical membrane potential measurements
If sodium pump activity and cytoskeletal proteins associated with ion regulation are altered, or ion channels are dysfunctional, or regulatory proteins are under expressed, then one might expect membrane potential of cells to be altered in pathologic mood states of bipolar patients. Indeed, examination of Transmembrane Potentials (TMP) in lymphocytes from manic type I patient’s reveals that the TMP of these cells is altered [45]. This is a state-related abnormality since while manic subjects have hyperpolarized circulating lymphocytes this membrane potential difference approaches normal with euthymia [45], and immortalized lymphoblasts of patients with bipolar illness and unaffected family members are not different from normal control subjects [46]. Lithium treatment would be expected to normalize the altered TMP [47], and may be responsible for normalization of intracellular ion concentrations [48]. These findings are important in their own right, but were also utilized as the basis for a potential diagnostic blood test for bipolar illness in which TMP is measured in ionically-stressed peripheral blood cells of symptomatic patients [49,50].
Imaging studies
Early PET imaging studies showed that whole brain glucose utilization was decreased in patients diagnosed with bipolar disorder compared with others [51]. Animal imaging studies demonstrated that animals that receive intracerebroventricular ouabain similarly have low levels of brain glucose utilization, but animals receiving ouabain with lithium pretreatment normalize glucose uptake [52]. Imaging studies also determined that the most abnormal glucose utilization was in the frontal lobe (the target of many antidepressant drugs) and the basal ganglia [53-55]. SPECT studies of the brain showed that patients with bipolar disorder have lower cerebral blood flow that is more evident in the frontal cortex and basal ganglia [56].
The sodium pump of the Na, K-ATPase utilizes about half of the brain’s total metabolic demand [57]. Therefore, low glucose and cerebral blood flow have direct effects on the function of the Na, K-ATPase and ion regulation in the brain.
Several different neurotransmitters have been implicated as being irregular through imaging studies in bipolar disorder. This includes dopamine [58-60], serotonin [61,62], GABA [63,64], and glutamate [65,66]. Because so many different neurotransmitters are irregular in bipolar disorder, it is logical to assume the pathogenesis is upstream from the individual neurotransmitters. The disorder most likely involves an irregularity before the synapse.
Animal models
Animal models are ultimately essential for confirming any hypothesis of pathogenesis of bipolar illness. Human studies can confirm associations and determine if predictions are indeed accurate, but can never produce cause-effect data, which are needed to determine that an abnormality causes a disease. Thus, while it may be difficult to know if an animal is “manic” or “depressed,” animal models are, nonetheless, the strongest evidence that can be provided. Several animal models demonstrate that alterations of ion homeostasis induce both manic-like behavior and depressive-like states in rodents.
A pharmacologic model in which ouabain, a potent inhibitor of brain-specific sodium pumps (α1 and α2 subunits of the Na, K-ATPase), is administered Intracerebroventricularly (ICV) to rats demonstrates that the rats will display for hyper- and hypo activity in a dose-related manner [67-69]. Lithium, and to some degree carbamazepine, reduce or normalize abnormal ouabain-induced behavioral changes [36,68]. The antipsychotics, haloperidol and cariprazine also normalize behavior in rats receiving ICV ouabain, and both agents are useful in mania [70-72]. This may be due to the activation of Na, K-ATPase activity by dopamine blockade [7].
Use of dihydro-ouabain in an in vitro model of stimulus-response in rat hippocampus slices is the only pre-clinical model for rapid cycling [8].
Genetic models have also been introduced. Knocking out the α3 isoform of the Na, K-ATPase produces behavior in rats that resembles both mania and depression and responds to treatments that may be helpful in patients with bipolar illness [10,73,74].
Downstream consequences of ion dysregulation
Increased intracellular sodium can alter other more active ions such as calcium and hydrogen (protons). Increases in free intracellular calcium as a function of mood state have been reported in peripheral blood cells of bipolar patients, including both platelets [4,75,76]and white blood cells [4]. Alterations in intracellular calcium may affect multiple second messenger systems [77,78]. This, in turn, can alter multiple cellular processes including excitability, mitochondrial activity, and resilience to apoptotic stimuli [79,80]. Additionally, there is an intimate relationship between elevated intracellular calcium and inflammation processes [81]; the latter has been associated with the pathophysiology of bipolar disorder [82].
Similarly, brain-imaging studies that measure intracellular pH have demonstrated a lower pH (i.e., higher proton concentrations) in unmediated bipolar adults [83,84] and manic adolescents [85]. Lithium and other mood stabilizers alkalize the cytoplasm [86,87]. These alterations may have a wide range of consequences in bipolar patients. Similarly, alterations in cytoplasmic pH will alter multiple aspects of neuronal function [88].
Both elevated intracellular calcium and hydrogen are targets of treatment in bipolar illness. Calcium channel blockers have been used successfully for the treatment of bipolar illness [89], but their use has not caught on for several reasons [90,91]. Similarly, reduced intracellular pH may respond to acidification of extracellular fluid as with the ketogenic diet [92], so that bipolar patients may achieve stability if they remain on the ketogenic diet [93].
Integrative model for pathogenesis of abnormal mood states
One of the mysteries of bipolar illness is the fact that the brain functions essentially normally in between episodes, but is quite impaired during an episode. While it is conceived as a mood disorder, it is clear that all aspects of brain function are impaired during an episode including motor movements, sensory perception, cognition, speed of response and processing, neuroendocrine function, and mood. Interepisode cognitive dysfunction is a late phenomenon, and probably occurs as a consequence of the neurotoxic aspects of mania and depression. Consequently, the essence of bipolar illness can be considered as episodic brain dysfunction. Any proposed hypothesis regarding the pathophysiology must take periods of normal brain function into account.
The studies reviewed in this paper suggest that a primary fault in ion transport or regulation can result in both manic and depressive symptoms. It is proposed that changes in resting membrane potential of both neurons and glia can produce both manic and depressive symptoms (Figure 1). These changes in membrane potential are found throughout and can actually be measured in ill patients. While the original hypothesis focused on sodium pump activity [21], changes in membrane potential can be brought about by changes in sodium channel, potassium channel, or sodium pump activity. These changes can be caused directly by abnormalities in the actual channels (such as the potassium channels), or indirectly by alterations in the cytoskeletal proteins supporting the channels (ankyrin G). The periodicity of this dysfunction may be related to rhythm control genes (CLOCK gene), or by ion transport regulating hormones that are inappropriately elaborated (endogenous cardenolides).
This hypothesis is consistent with treatment response. Nearly all effective treatments used in bipolar illness reduce the concentration of sodium, calcium, or hydrogen either directly or indirectly [6,7]. The most effective mood stabilizers are agents that reduce intracellular sodium accumulation in an activity-dependent fashion (i.e., lithium, valproic acid, carbamazepine, and lamotrigine).
Nonetheless, there is great heterogeneity in the presentation, genetics, and treatment response in people with bipolar disorder. This suggests the presence of endophenotypes in this condition. Ongoing research examining the ion regulatory apparati in bipolar disorder is required to clarify the actual pathoetiologic mechanisms.
References
- Goodwin FK, Jamison KR. Manic-depressive illness: Bipolar Disorders and Recurrent Depression.2nd Edition. New York: Oxford University Press. 2007.
- Askland KD. Editorial: "Ion channels and mental illness: exploring etiology and pathophysiology in major psychiatric disorders". Front Genet. 2015; 6: 152.
- Coppen A, Shaw DM, Malleson A, Costain R. Mineral metabolism in mania. Br Med J. 1966; 1: 71-75.
- Dubovsky SL, Murphy J, Thomas M, Rademacher J. Abnormal intracellular calcium ion concentration in platelets and lymphocytes of bipolar patients. Am J Psychiatry. 1992; 149: 118-120.
- Goldstein I, Lerer E, Laiba E, Mallet J, Mujaheed M, Laurent C, et al. Association between sodium- and potassium-activated adenosine triphosphatase alpha isoforms and bipolar disorders. Biol Psychiatry. 2009; 65: 985-991.
- El-Mallakh RS, Huff MO. Mood stabilizers and ion regulation. Harv Rev Psychiatry. 2001; 9: 23-32.
- Roberts RJ, Repass R, El-Mallakh RS. Effect of dopamine on intracellular sodium: a common pathway for pharmacological mechanism of action in bipolar illness. World J Biol Psychiatry. 2010; 11: 181-187.
- El-Mallakh RS, Schurr A, Payne RS, Li R. Ouabain induction of cycling of multiple spike responses in hippocampal slices is delayed by lithium. J Psychiatr Res. 2000; 34: 115-120.
- El-Mallakh RS, El-Masri MA, Huff MO, Li XP, Decker S, Levy RS. Intracerebroventricular administration of ouabain as a model of mania in rats. Bipolar Disord. 2003; 5: 362-365.
- Kirshenbaum GS, Clapcote SJ, Duffy S, Burgess CR, Petersen J, Jarowek KJ, et al. Mania-like behavior induced by genetic dysfunction of the neuron-specific Na+,K+-ATPase α3 sodium pump. Proc Natl Acad Sci U S A. 2011; 108: 18144-18149.
- Cade JF. Lithium salts in the treatment of psychotic excitement. Med J Aust. 1949; 2: 349-352.
- Hodgkin AL, Huxley AF. Propagation of electrical signals along giant nerve fibers. Proc R Soc Lond B Biol Sci. 1952; 140: 177-183.
- Linderholm H. On the behavior of the sodium pump in from skin at various concentrations of Na ions in the solution on the epithelial side. Acta Physiol Scand. 1954; 31: 36-61.
- Naylor GJ, McNamee HB, Moody JP. Erythrocyte sodium and potassium in depressive illness. J Psychosom Res. 1970; 14: 173-177.
- Naylor GJ, McNamee HB, Moody JP. Changes in erythrocyte sodium and potassium on recovery from a depressive illness. Br J Psychiatry. 1971; 118: 219-223.
- Shaw DM. Mineral metabolism, mania, and melancholia. Br Med J. 1966; 2: 262-267.
- Hokin-Neaverson M, Spiegel DA, Lewis WC. Deficiency of erythrocyte sodium pump activity in bipolar manic-depressive psychosis. Life Sci. 1974; 15: 1739-1748.
- Nurnberger JJr, Jimerson DC, Allen JR, Simmons S, Gershon E. Red cell ouabain-sensitive Na+-K+-adenosine triphosphatase: a state marker in affective disorder inversely related to plasma cortisol. Biol Psychiatry. 1982; 17: 981-992.
- Naylor GJ, Smith AH. Defective genetic control of sodium-pump density in manic depressive psychosis. Psychol Med. 1981; 11: 257-263.
- Looney SW, el-Mallakh RS. Meta-analysis of erythrocyte Na,K-ATPase activity in bipolar illness. Depress Anxiety. 1997; 5: 53-65.
- el-Mallakh RS, Wyatt RJ. The Na, K-ATPase hypothesis for bipolar illness. Biol Psychiatry. 1995; 37: 235-244.
- Ferreira MA, O'Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. 2008; 40: 1056-1058.
- Schulze TG, Detera-Wadleigh SD, Akula N, Gupta A, Kassem L, Steele J, et al. Two variants in Ankyrin 3 (ANK3) are independent genetic risk factors for bipolar disorder. Mol Psychiatry. 2009; 14: 487-491.
- Smith EN, Bloss CS, Badner JA, Barrett T, Belmonte PL, Berrettini W, et al. Genome-wide association study of bipolar disorder in European American and African American individuals. Mol Psychiatry. 2009; 14: 755-763.
- Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, Craddock N, et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011; 43: 977–983.
- Borsotto M, Cavarec L, Bouillot M, Romey G, Macciardi F, Delaye A, et al. PP2A-Bgamma subunit and KCNQ2 K+ channels in bipolar disorder. Pharmacogenomics J. 2007; 7: 123-132.
- Judy JT, Seifuddin F, Pirooznia M, Mahon PB, Jancic D, Goes FS, et al. Converging Evidence for Epistasis between ANK3 and Potassium Channel Gene KCNQ2 in Bipolar Disorder. Front Genet. 2013; 4: 87.
- Kaminsky Z, Jones I, Verma R, Saleh L, Trivedi H, Guintivano J, et al. DNA methylation and expression of KCNQ3 in bipolar disorder. Bipolar Disord. 2015; 17: 150-159.
- Uemura T, Green M, Warsh JJ. CACNA1C SNP rs1006737 associates with bipolar I disorder independent of the Bcl-2 SNP rs956572 variant and its associated effect on intracellular calcium homeostasis. World J Biol Psychiatry. 2015: 1-10.
- Cooper EC. Made for "anchorin": Kv7.2/7.3 (KCNQ2/KCNQ3) channels and the modulation of neuronal excitability in vertebrate axons. Semin Cell Dev Biol. 2011; 22: 185-192.
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005; 6: 850-862.
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005; 8: 51-60.
- Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, et al. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci USA. 1991; 88: 6259-6263.
- Weinberg U, Dolev S, Shapiro MS, Shilo L, Rabinowitz R, Shenkman L. Digoxinlike immunoreactive factor isolated from human pleural fluid is structurally similar to digoxin. Am J Med Sci. 1997; 314: 28-30.
- Sophocleous A, Elmatzoglou I, Souvatzoglou A. Circulating endogenous digitalis-like factor(s) (EDLF) in man is derived from the adrenals and its secretion is ACTH-dependent. J Endocrinol Invest. 2003; 26: 668-674.
- El-Mallakh RS, Miller J, Valdes R Jr, Cassis TB, Li R. Digoxin-like immunoreactive factor in human cerebrospinal fluid. J Neuropsychiatry Clin Neurosci. 2007; 19: 91.
- Balzan S, D'Urso G, Nicolini G, Forini F, Pellegrino M, Montali U. Erythrocyte sodium pump stimulation by ouabain and an endogenous ouabain-like factor. Cell Biochem Funct. 2007; 25: 297-303.
- Weiss DN, Podberesky DJ, Heidrich J, Blaustein MP. Nanomolar ouabain augments caffeine-evoked contractions in rat arteries. Am J Physiol. 1993; 265: C1443-1448.
- El-Mallakh RS, Stoddard M, Jortani SA, El-Masri MA, Sephton S, Valdes R Jr. Aberrant regulation of endogenous ouabain-like factor in bipolar subjects. Psychiatry Res. 2010; 178: 116-120.
- Valdes R, Hagberg JM, Vaughan TE, Lau BW, Seals DR, Ehsani AA. Endogenous digoxin-like immunoreactivity in blood is increased during prolonged strenuous exercise. Life Sci. 1988; 42: 103-110.
- Shah A, Alshaher M, Dawn B, Siddiqui T, Longaker RA, Stoddard MF, et al. Exercise tolerance is reduced in bipolar illness. J Affect Disord. 2007; 104: 191-195.
- Grider G, El-Mallakh RS, Huff MO, Buss TJR, Miller J, Valdes R. Endogenous digoxin-like immunoreactive factor (DLIF) serum concentrations are decreased in manic bipolar patients compared to normal controls. J Affect Disord. 1999; 54: 261-267.
- Jacobs BE, Liu Y, Pulina MV, Golovina VA, Hamlyn JM. Normal pregnancy: mechanisms underlying the paradox of an ouabain-resistant state with elevated endogenous ouabain, suppressed arterial sodium calcium exchange, and low blood pressure. Am J Physiol Heart Circ Physiol. 2012; 302: H1317-H1329.
- Vakkuri O, Arnason SS, Pouta A, Vuolteenaho O, Leppäluoto J. Radioimmunoassay of plasma ouabain in healthy and pregnant individuals. J Endocrinol. 2000; 165: 669-677.
- El-Mallakh RS, Li R, Worth CA, Peiper SC. Leukocyte transmembrane potential in bipolar illness. J Affect Disord. 1996; 41: 33-37.
- Buss TJ, Li R, Peiper SC, El-Mallakh RS. Lymphoblastoid transmembrane potential in bipolar patients, their siblings, and unrelated healthy comparison subjects. Psychiatry Res. 1996; 59: 197-201.
- Thiruvengadam A. Effect of lithium and sodium valproate ions on resting membrane potentials in neurons: an hypothesis. J Affect Disord. 2001; 65: 95-99.
- Huang X, Lei Z, El-Mallakh RS. Lithium normalizes elevated intracellular sodium. Bipolar Disord. 2007; 9: 298-300.
- Thiruvengadam AP, Chandrasekaran K. Evaluating the validity of blood-based membrane potential changes for the identification of bipolar disorder I. J Affect Disord. 2007; 100: 75-82.
- Woodruff DB, El-Mallakh RS, Thiruvengadam AP. Validation of a diagnostic screening blood test for bipolar disorder. Ann Clin Psychiatry. 2012; 24: 135-139.
- Baxter LR, Phelps ME, Mazziotta JC, Schwartz JM, Gerner RH, Selin CE, et al. Cerebral metabolic rates for glucose in mood disorders. Studies with positron emission tomography and fluorodeoxyglucose F 18. Arch Gen Psychiatry. 1985; 42: 441-447.
- Hougland MT, Gao Y, Herman L, Ng CK, Lei Z, El-Mallakh RS. Positron emission tomography with fluorodeoxyglucose-F18 in an animal model of mania. Psychiatry Research: Neuroimaging. 2008; 164: 166-171.
- Buchsbaum MS. Brain imaging in the search for biological markers in affective disorder. J Clin Psychiatry. 1986; 47: 7-12.
- Buchsbaum MS, Wu J, DeLisi LE, Holcomb H, Kessler R, Johnson J, et al. Frontal cortex and basal ganglia metabolic rates assessed by positron emission tomography with [18F]2-deoxyglucose in affective illness. J Affect Disord. 1986; 10: 137-152.
- Cohen RM, Semple WE, Gross M, Nordahl TE, King AC, Pickar D, et al. Evidence for common alterations in cerebral glucose metabolism in major affective disorders and schizophrenia. Neuropsychopharmacology. 1989; 2: 241-254.
- O'Connell RA, Van Heertum RL, Luck D, Yudd AP, Cueva JE, Billick SB, et al. Single-photon emission computed tomography of the brain in acute mania and schizophrenia. J Neuroimaging. 1995; 5: 101-104.
- Astrup J, Sørensen PM, Sørensen HR. Oxygen and glucose consumption related to Na+-K+ transport in canine brain. Stroke. 1981; 12: 726-730.
- Ames A. CNS energy metabolism as related to function. Brain Res Brain Res Rev. 2000; 34: 42-68.
- Suhara T, Nakayama K, Inoue O, Fukuda H, Shimizu M, Mori A, et al. D1 dopamine receptor binding in mood disorders measured by positron emission tomography. Psychopharmacology. 1992; 106: 14-18.
- Vaughan RA, Foster JD. Mechanisms of dopamine transporter regulation in normal and disease states. Trends Pharmacol Sci. 2013; 34: 489-496.
- Sullivan GM, Ogden RT, Oquendo MA, Kumar JS, Simpson N, Huang YY, et al. Positron emission tomography quantification of serotonin-1A receptor binding in medication-free bipolar depression. Biol Psychiatry. 2009; 66: 223-230.
- Hsu JW, Lirng JF, Wang SJ, Lin CL, Yang KC, Liao MH, et al. Association of thalamic serotonin transporter and interleukin-10 in bipolar I disorder: a SPECT study. Bipolar Disord. 2014; 16: 241-248.
- Torrey EF, Barci BM, Webster MJ, Bartko JJ, Meador-Woodruff JH, Knable MB. Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol Psychiatry. 2005; 57: 252-260.
- Brady RO, McCarthy JM, Prescot AP, Jensen JE, Cooper AJ, Cohen BM, et al. Brain gamma-aminobutyric acid (GABA) abnormalities in bipolar disorder. Bipolar Disord. 2013; 15: 434-439.
- Castillo M, Kwock L, Courvoisie H, Hooper SR. Proton MR spectroscopy in children with bipolar affective disorder: preliminary observations. AJNR Am J Neuroradiol. 2000; 21: 832-838.
- Xu J, Dydak U, Harezlak J, Nixon J, Dzemidzic M, Gunn AD, et al. Neurochemical abnormalities in unmedicated bipolar depression and mania: a 2D 1H MRS investigation. Psychiatry Res. 2013; 213: 235-241.
- el-Mallakh RS, Harrison LT, Li R, Changaris DG, Levy RS. An animal model for mania: preliminary results. Prog Neuropsychopharmacol Biol Psychiatry. 1995; 19: 955-962.
- Li R, El-Mallakh RS, Harrison L, Changaris DG, Levy RS. Lithium prevents ouabain-induced behavioral changes: Toward an animal model for manic-depression. Mol Chem Neuropathol. 1997; 31: 65-72.
- Wang YC, Wang EN, Wang CC, Huang CL, Huang AC. Effects of lithium and carbamazepine on spatial learning and depressive behavior in a rat model of bipolar disorder induced by ouabain. Pharmacol Biochem Behav. 2013; 105: 118-127.
- El-Mallakh RS, Payne RS, Schurr A, Gao Y, Lei Z, Kiss B, et al. Cariprazine delays ouabain-evoked epileptiform spikes and loss of activity in rat hippocampal slices. Psychiatry Res. 2015; 229: 370-373.
- El-Mallakh RS, Decker S, Morris M, Li XP, Huff MO, El-Masri MA, et al. Efficacy of olanzapine and haloperidol in an animal model of mania. Prog Neuropsychopharmacol Biol Psychiatry. 2006; 30: 1261-1264.
- Gao Y, Peterson S, Masri B, Hougland MT, Adham N, Gyertyán I, et al. Cariprazine exerts antimanic properties and interferes with dopamine D2 receptor ß-arrestin interactions. Pharmacol Res Perspect. 2015; 3: e00073.
- Kirshenbaum GS, Clapcote SJ, Petersen J, Vilsen B, Ralph MR, Roder JC. Genetic suppression of agrin reduces mania-like behavior in Na+, K+ -ATPase α3 mutant mice. Genes Brain Behav. 2012; 11: 436-443.
- Kirshenbaum GS, Burgess CR, Déry N, Fahnestock M, Peever JH, Roder JC. Attenuation of mania-like behavior in Na(+),K(+)-ATPase α3 mutant mice by prospective therapies for bipolar disorder: melatonin and exercise. Neuroscience. 2014; 260: 195-204.
- Dubovsky SL, Lee C, Christiano J, Murphy J. Elevated platelet intracellular calcium concentration in bipolar depression. Biol Psychiatry. 1991; 29: 441-450.
- Dubovsky SL, Daurignac E, Leonard KE. Increased platelet intracellular calcium ion concentration is specific to bipolar disorder. J Affect Disord. 2014; 164: 38-42.
- Uemura T, Green M, Corson TW, Perova T, Li PP, Warsh JJ. Bcl-2 SNP rs956572 associates with disrupted intracellular calcium homeostasis in bipolar I disorder. Bipolar Disord. 2011; 13: 41-51.
- Hayashi A, Le Gal K, Sodersten K, Vizlin-Hodzic D, Agren H, Funa K. Calcium-dependent intracellular signal pathways in primary cultured adipocytes and ANK3 gene variation in patients with bipolar disorder and healthy controls. Mol Psychiatry. 2015; 20: 931-940.
- Gao Y, Lei Z, Lu C, Roisen FJ, El-Mallakh RS. Effect of ionic stress on apoptosis and the expression of TRPM2 in human olfactory neuroepithelial-derived progenitors. World J Biol Psychiatry. 2010; 11: 972-984.
- Ament SA, Szelinger S, Glusman G, Ashworth J, Hou L, Akula N, et al. Rare variants in neuronal excitability genes influence risk for bipolar disorder. Proc Natl Acad Sci USA. 2015; 112: 3576-3581.
- Scheff NN, Gold MS. Trafficking of Na+/Ca2+ exchanger to the site of persistent inflammation in nociceptive afferents. J Neurosci. 2015; 35: 8423-8432.
- Fiedorowicz JG, Prossin AR, Johnson CP, Christensen GE, Magnotta VA, Wemmie JA. Peripheral inflammation during abnormal mood states in bipolar I disorder. J Affect Disord. 2015; 187: 172-178.
- Kato T, Murashita J, Kamiya A, Shioiri T, Kato N, Inubushi T. Decreased brain intracellular pH measured by 31P-MRS in bipolar disorder: a confirmation in drug-free patients and correlation with white matter hyperintensity.Eur Arch Psychiatry ClinNeurosci. 1998; 248: 301-306.
- Hamakawa H, Murashita J, Yamada N, Inubushi T, Kato N, Kato T. Reduced intracellular pH in the basal ganglia and whole brain measured by 31P-MRS in bipolar disorder. Psychiatry Clin Neurosci. 2004; 58: 82-88.
- Weber WA, Dudley J, Lee JH, Strakowski SM, Adler CM, DelBello MP. A pilot study of alterations in high energy phosphoryl compounds and intracellular pH in unmedicated adolescents with bipolar disorder. J Affect Disord. 2013; 150: 1109-1113.
- Song D, Du T, Li B. Astrocyticalkalinization by therapeutically relevant lithium concentrations: implications for myo-inositol depletion Psychopharmacology (Berl). 2008; 200: 187-195.
- Song D, Li B, Yan E, Cai L, Gu L, Li H, et al. Chronic treatment with anti-bipolar drugs causes’ intracellular alkalinization in astrocytes, altering their functions. Neurochem Res. 2012; 37: 2524-2540.
- Ruffin VA, Salameh AI, Boron WF, Parker MD. Intracellular pH regulation by acid-base transporters in mammalian neurons. Front Physiol. 2014; 5: 43.
- Dubovsky SL. Calcium antagonists in manic-depressive illness. Neuropsychobiology. 1993; 27: 184-192.
- Dubovsky SL. Why don't we hear more about the calcium antagonists? An industry-academia interaction. Biol Psychiatry. 1994; 35: 149-150.
- Casamassima F, Hay AC, Benedetti A, Lattanzi L, Cassano GB, Perlis RH. L-type calcium channels and psychiatric disorders: A brief review. Am J Med Genet B Neuropsychiatr Genet. 2010; 153B: 1373-1390.
- El-Mallakh RS, Paskitti ME. The ketogenic diet may have mood-stabilizing properties. Med Hypotheses. 2001; 57: 724-726.
- Phelps JR, Siemers SV, El-Mallakh RS. The ketogenic diet for type II bipolar disorder. Neurocase. 2013; 19: 423-426.