
Review Article
J Immun Res. 2014;1(2): 4.
CD1d-restricted Natural Killer T Cells in Metabolic Disorders
Tajiri K1*
1Department of Gastroenterology, Toyama University Hospital, Japan
*Corresponding author: Tajiri K, Department of Gastroenterology, Toyama University Hospital, Japan
Received: September 22, 2014; Accepted: October 16, 2014; Published: October 20, 2014
Abstract
Natural killer T (NKT) cells are a unique subset of innate immune cells, which express both T cell receptors (TCRs) and natural killer receptors. Among NKT cells, invariant NKT (iNKT) cells express in variant TCRs, recognize glycolipid antigens presented by CD1d, and produce both pro inflammatory and anti inflammatory cytokines. Recent findings showed that NKT cells, especially iNKT cells, play a regulatory role in metabolic abnormalities mainly through the interaction with CD1d. This review focused on NKT cells in obesity and their contribution to metabolic disorders, such as glucose intolerance, atherosclerosis, and non alcoholic fatty liver disease.
Keywords: NKT cell; CD1d; Metabolic abnormality; Glycolipid antigen; MTP
Introduction
Obesity has become a major problem around the world due to its contribution to morbidity and mortality. Obesity is one of the essential contributors to the development of metabolic diseases, such as hypertension, hyperlipidemia, atherosclerosis, and diabetes mellitus [1]. In addition, obesity is thought to be closely associated with systemic inflammation, which is an underlying contributor to many of these metabolic diseases [2, 3]. Natural killer T cells are innate immune cells, can acts either pro- or anti-inflammatory, and work as immune regulatory. Invariant natural killer T (iNKT) cells are a unique NKT cell population expressing invariant T cell receptors (TCRs) (Va24 in humans and Va14 in mice), recognize glycolipid antigens through CD1d, and can be either pro- or antiinflammatory [4]. The subsets of NKT cells are found preferentially in the liver [5] and adipose tissue [6, 7], which are essential sites of lipid metabolic regulation, and are considered essential participants in metabolic abnormalities. This review summarizes and discusses the role of CD1d-restricted NKT cells in obesity and their contribution to metabolic disorders.
NKT Cells in Lipid Metabolism
Obesity develops as the result of increases in adipose tissues due to excessive energy intake. White adipose tissues is the primary site of energy storage, and white fat storage is associated with the metabolic complications of obesity [8]. On the other hand, microsomal triglyceride transfer protein (MTP) mainly located in the endoplasmic reticulum (ER) of hepatocytes and intestinal epithelial cells plays an essential role in the transfer of lipids, including phospholipids, triglycerides, and cholesterol. Dietary lipids are taken up by the enterocytes through various transporters as free fatty acids, monoacylglycerols and free cholesterol, transferred to the ER by MTP, and used for the synthesis of phospholipids, triacylglycerols and cholesterol esters [9]. These lipids are stored in the cytosol as lipid droplets. MTP is a key protein in the assembly and secretion of triglyceride-rich lipoproteins (apolipoprotein B, apoB) in the intestine and liver, and the abnormality of such lipoproteins predispose individuals to various metabolic diseases, such as obesity, atherosclerosis, diabetes, and nonalcoholic fatty liver disease [10].
In addition, iNKT cells are lipid antigen-specific lymphocytes that recognize glycolipid antigens presented on CD1d molecules and produce large amounts of T helper (Th)1 cytokines, including interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-a); and Th2 cytokines, including interleukins (IL)-4 and IL-10 [4]. CD1d is a major histocompatibility complex (MHC) class 1-like molecule widely found in systemic organs [11, 12]. The function of glycolipid antigen presentation by CD1d has been shown to be regulated by MTP [13]. MTP deficiency is associated with impaired activation and reduced number and phenotypic alterations of NKT cells, resulting in resistance to immune pathogenesis associated with NKT cellmediated diseases [13 – 15]. The regulation of CD1d function by MTP occurred not only in apoB-secreting cells, such as hepatocytes and intestinal epithelial cells (IECs) [13], but also antigen presenting cells (APCs) [14, 15] and adipocyte [16]. The precise function of MTP in adipocytes or APCs is unknown. MTP is presumed to load the first endogenous lipid into the CD1d in APCs, where it was suggested to play a role in lipid droplet formation [16]. Moreover, NKT cell-mediated inflammation was recently shown to be regulated by interactions among CD1d, MTP, and cytokines [17]. Thus, MTP is an essential protein not only in lipid metabolism but also the lipid antigen presentation function of CD1d.
Furthermore, proteins involved in lipoprotein metabolism other than MTP, such as low-density lipoprotein receptor (LDLr) [18, 19], scavenger receptors [20], and cholesterol membrane transporters [21], are able to modulate NKT cells homeostasis and activation. LDLr-related protein has been shown to be expressed in macrophages and to be necessary for the production of Th2 cytokines not but Th1 cytokines [19]. These data suggest that not only the modification of antigen presentation on CD1d by MTP but also lipid transfer and metabolism by lipid receptors may affect the functions of NKT cells.
NKT Cells and Intestinal Microbiota
Innate immune cells, including NKT cells, can also be activated by intestinal microbiota-derived antigens through Toll-like receptor (TLR) signaling [22 – 24]. Recently, commensally microbiota was also shown to regulate the development and function of CD1drestricted NKT cells through interactions with lipid antigens [25- 27]. Intestinal microbiota-derived lipids and metabolites, as well as cytokines and chemokines produced in response to microbial recognition, may contribute to systemic NKT cell development [28]. The intestinal microbiota is different between healthy and obese subjects [29]. The relative proportion of the fecal microbiota are also altered in obese human subjects but change with weight loss [30]. Furthermore, transplantation of intestinal microbiota from obese mice resulted in greater adiposity in recipients than transplantation of microbiota from lean donors [31]. These differences in the intestinal microbiota associated with obesity may affect the function of NKT cells. Probiotic antigens may stimulate hepatic NKT cells and restore the number of hepatic NKT cells in mice fed a high-fat diet through interactions between lipid antigens and CD1d, but not through TLR4 signaling [32]. Thus, NKT cells are regulated by not only glycolipid antigen presentation through CD1d but also intestinal microbiotaderived factors. NKT cells may be associated with the development of systemic inflammation through metabolic modifications from lean to obese.
The Contribution of NKT Cells to Metabolic Abnormalities
a. Obesity
Recent studies have suggested that NKT cells are involved in systemic metabolic disorders. For example, iNKT cells were found to be depleted in the adipose tissue of obese individuals [6, 7, 33], while restoring iNKT cells by adoptive transfer induced weight loss [7]. This iNKT cell depletion was correlated with proinflammatory macro phage infiltration [7]. The numbers of iNKT cells could be restored after weight loss [7]. CD1d-deficient mice fed a high-fat diet have been shown to aggravate metabolic parameters, such as glucose homeostasis and hepatic lipid metabolism [34]. Furthermore, CD1ddeficient mice fed a high-fat diet were more susceptible to weight gain, along with increased adiposity and greater induction of inflammatory gene expression in the liver and white adipose tissues [35]. These findings suggest that CD1d-restricted NKT cells protect against dietinduced obesity through the regulation of cytokine production [36].
On the other hand, obesity-induced inflammation is known to induce various metabolic disorders [37]. CD1d is expressed at high levels in adipocytes and CD1d-expressing adipocytes regulate iNKT cells. The iNKT cell population and CD1d expression level were shown to be reduced in the adipose tissues of obese mice and humans, and iNKT cell-deficient mice became more obese and exhibited increased adipose tissue inflammation at an early stage of obesity [38]. Furthermore, lack of iNKT cells was shown to affect lipid metabolism in the adipose tissue of diet-induced obese mice [39]. Ja18-deficent mice, which lack iNKT cells, were resistant to diet-induced obesity and showed increased lipogenesis counter balanced by elevated lipase expansion and basal lipolysis [39].
Adoptive transfer of iNKT cells and a-galactosylceramide treatment were shown to protect against weight gain and adipocyte hypertrophy and to reverse obesity-associated metabolic disorders [7]. Thus, iNKT cells are associated with obesity and obesity-induced metabolic disorders, and may be a potential therapeutic target for obesity-induced metabolic disorders.
b. Glucose intolerance
CD1d-deficient mice, which lack iNKT cells, were shown to worsen glucose homeostasis when they had been fed a high-fat diet [34, 35], while adoptive transfer of iNKT cells improved glucose intolerance [7]. Adoptive transfer of iNKT cells or treatment with glycolipid antigens has been shown to improve glucose intolerance in ob/ob mice, which are deficient in leptin and are regarded as a model for obesity [40, 41]. CD1d-restricted iNKT cells in adipose tissue were shown to play an essential role in preventing insulin resistance [42]. NKT cell function was directly modulated by adipocytes, which acted as lipid antigen presenting cells in a CD1d-dependent manner [42]. These findings suggest that CD1d-restricted iNKT cells protect against glucose intolerance [36].
c. Atherosclerosis
Atherosclerosis is one of the major disorders due to metabolic abnormalities, which leads to induce various serious complications. CD1d-restricted iNKT cells were shown to exacerbate atherosclerosis through the production of proinflammatory cytokines [43, 44]. Atherosclerogenic ApoE-deficient mice crossed with CD1d-deficient mice lacking iNKT cells were shown to have a 25% reduction in atherosclerosis lesion size [43]. Furthermore, ApoE-deficient mice treated with a-galactosylceramide, a glycolipid that activates iNKT cells, showed a 50% increase in atherosclerosis with inflammatory Th1 and Th2 cytokines [43]. Increasing the complement of iNKT cells exacerbated aortic atherosclerosis and inflammation in obesogenic diet-fed LDLr-deficient mice, which are susceptible to dyslipidemia, hyperinsulinemia, insulin resistance, and hepatic triglyceride accumulation [45]. Furthermore, a-galactosylceramide treatment of ApoE-deficient mice with established atherosclerosis lesions had no significant effect on lesion size, but decreased their collagen content in atherosclerosis [46]. Thus, CD1d-restricted iNKT cells play a role in the formation of atherosclerosis through proinflammatory factors.
d. Nonalcoholic fatty liver disease (NAFLD)
CD1d-deficient mice, which lack iNKT cells, fed a high-fat diet showed increased susceptibility to fatty liver, along with increased adiposity and greater induction of inflammatory genes in the liver compared to normal controls [35]. Depletion of NKT cells has also been reported in ob/ob mice, which are regarded as a model of obesity-related fatty liver, through hepatic sensitization toward proinflammatory conditions induced by endotoxins from the gut, by increased production of adipokines, or by ER stress [47 – 50]. In wild-type mice fed a choline-deficient or high-fat diet, which are also regarded as a model of NAFLD, reductions in the numbers of hepatic NKT cells were accompanied by increased Th1 cytokine production [51, 52]. Hepatic NKT cells were also reported to be decreased in hepatosteatosis through KC- [53] and IL-12 [54]-dependent mechanisms. In addition, administration of probiotics has been reported to improve high-fat diet-induced hepatic steatosis by increasing hepatic NKT cells through reductions in TNF-a production and nuclear factor–κB binding activity [55].
Adoptive transfer of NKT cells or treatment with glycolipid antigens has been shown to reduce hepatic steatosis in ob/ob mice [40, 41]. Moreover, adrenergic activation by norepinephrine has been reported to induce the expansion of NKT cell populations and improve hepatic steatosis [48]. In humans, NKT cell numbers were decreased in the livers of patients with relatively mild NAFLD [54]. Taken together, these findings indicate that hepatic NKT cells are preferentially protective during the process of hepatic steatosis through various metabolic factors and cytokines, especially those produced by KCs and associated with gut-derived factors, such as endotoxin.
During advanced stages of NAFLD, the number of hepatic NKT cells was increased in the liver. These increases were accompanied by increased activation of the hedgehog (Hh) pathway and increased osteopontin production, leading to promotion of liver fibrosis through activation of hepatic stellate cells [56, 57]. In human NAFLD, the number of hepatic NKT cells increases with disease progression [58, 59]. Furthermore, disease progression is accompanied by increased activation of antigen presenting cells, such as KCs, and increased expression of CD1d [58]. Thus, CD1d-restricted NKT cells are activated in the livers of patients with NAFLD, at least in those with advanced disease.
Summary and Conclusion
CD1d-restricted NKT cells may play an essential role in the interaction between metabolism and the immune system to regulate energy balance through factors derived from the gut (Figure 1). Antigen presentation of CD1d through the interaction of lipidderived factors and sensitization of NKT cells from gut-derived factors through macrophages may have important roles in metabolic disorders. The modulation of NKT cells may be a therapeutic target in various metabolic diseases.
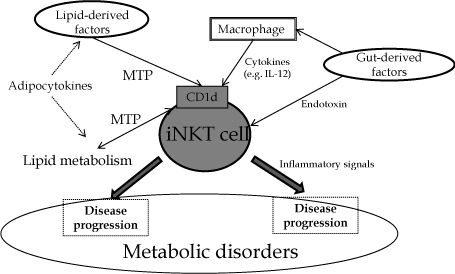
Figure 1: Schema of interactions of NKT cells in metabolic disorders. MTP: microsomal triglyceride transfer protein.
References
- Moller DE, Kaufman KD . Metabolic syndrome: a clinical and molecular perspective. Annu Rev Med. 2005; 56: 45-62.
- Hotamisligil GS . Inflammation and metabolic disorders. Nature. 2006; 444: 860-867.
- Gregor MF, Hotamisligil GS . Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011; 29: 415-445.
- Godfrey DI, Kronenberg M . Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest. 2004; 114: 1379-1388.
- Mehal WZ, Azzaroli F, Crispe IN . Immunology of the healthy liver: old questions and new insights. Gastroenterology. 2001; 120: 250-260.
- Lynch L, O'Shea D, Winter DC, Geoghegan J, Doherty DG, O'Farrelly C . Invariant NKT cells and CD1d(+) cells amass in human omentum and are depleted in patients with cancer and obesity. Eur J Immunol. 2009; 39: 1893-1901.
- Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V, et al . Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity. 2012; 37: 574-587.
- Gesta S, Tseng YH, Kahn CR . Developmental origin of fat: tracking obesity to its source. Cell. 2007; 131: 242-256.
- Hussain MM . Intestinal lipid absorption and lipoprotein formation. Curr Opin Lipidol. 2014; 25: 200-206.
- Cuchel M, Rader DJ . Microsomal transfer protein inhibition in humans. Curr Opin Lipidol. 2013; 24: 246-250.
- Porcelli SA, Modlin RL . The CD1 system: antigen-presenting molecules for T cell recognition of lipids and glycolipids. Annu Rev Immunol. 1999; 17: 297-329.
- Bilsland CA, Milstein C . The identification of the beta 2-microglobulin binding antigen encoded by the human CD1D gene. Eur J Immunol. 1991; 21: 71-78.
- Brozovic S1, Nagaishi T, Yoshida M, Betz S, Salas A, Chen D, et al . CD1d function is regulated by microsomal triglyceride transfer protein. Nat Med. 2004; 10: 535-539.
- Dougan SK, Rava P, Hussain MM, Blumberg RS . MTP regulated by an alternate promoter is essential for NKT cell development. J Exp Med. 2007; 204: 533-545.
- Zeissig S, Dougan SK, Barral DC, Junker Y, Chen Z, Kaser A, et al . Primary deficiency of microsomal triglyceride transfer protein in human abetalipoproteinemia is associated with loss of CD1 function. J Clin Invest. 2010; 120: 2889-2899.
- Rakhshandehroo M, Gijzel SM, Siersbaek R, Broekema MF, de Haar C, Schipper HS, et al. CD1d-mediated presentation of endogenous lipid antigens by adipocytes requires microsomal triglyceride transfer protein (MTP). J Biol Chem 2014; 289: 22128-22139.
- Olszak T, Neves JF1, Dowds CM2, Baker K3, Glickman J4, Davidson NO5, et al . Protective mucosal immunity mediated by epithelial CD1d and IL-10. Nature. 2014; 509: 497-502.
- van den Elzen P, Garg S, León L, Brigl M, Leadbetter EA, Gumperz JE, et al. Apolipoprotein-mediated pathways of lipid antigen presentation. Nature. 2005; 437: 906-910.
- Covarrubias R, Wilhelm AJ2, Major AS3 . Specific deletion of LDL receptor-related protein on macrophages has skewed in vivo effects on cytokine production by invariant natural killer T cells. PLoS One. 2014; 9: e102236.
- Freigang S, Landais E, Zadorozhny V, Kain L, Yoshida K, Liu Y, et al . Scavenger receptors target glycolipids for natural killer T cell activation. J Clin Invest. 2012; 122: 3943-3954.
- Sag D, Wingender G, Nowyhed H, Wu R, Gebre AK, Parks JS, et al. ATP-binding cassette transporter G1 intrinsically regulates invariant NKT cell development. J Immunol. 2012; 189: 5129-5138.
- Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB . Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat Immunol. 2003; 4: 1230-1237.
- Nagarajan NA, Kronenberg M . Invariant NKT cells amplify the innate immune response to lipopolysaccharide. J Immunol. 2007; 178: 2706-2713.
- Brigl M, Tatituri RV, Watts GF, Bhowruth V, Leadbetter EA, Barton N, et al. Innate and cytokine-driven signals, rather than microbial antigens, dominate in natural killer T cell activation during microbial infection. J Exp Med 2011; 208:1163-1177.
- An D, Oh SF1, Olszak T2, Neves JF2, Avci FY3, Erturk-Hasdemir D1, et al . Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell. 2014; 156: 123-133.
- Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, et al . Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012; 336: 489-493.
- Wingender G, Stepniak D, Krebs P, Lin L, McBride S, Wei B, et al . Intestinal microbes affect phenotypes and functions of invariant natural killer T cells in mice. Gastroenterology. 2012; 143: 418-428.
- Zeissig S, Blumberg RS2 . Commensal microbial regulation of natural killer T cells at the frontiers of the mucosal immune system. FEBS Lett. 2014; .
- Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI . Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005; 102: 11070-11075.
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI . Microbial ecology: human gut microbes associated with obesity. Nature. 2006; 444: 1022-1023.
- Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI . Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A. 2007; 104: 979-984.
- Liang S, Webb T, Li Z . Probiotic antigens stimulate hepatic natural killer T cells. Immunology. 2014; 141: 203-210.
- Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KS, et al. Activation of natural killer T cells promotes M2 Macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4 (IL-4)/STAT6 protein signaling axis in obesity. J Biol Chem 2012; 287:13561-13571.
- Kotas ME, Lee HY, Gillum MP, Annicelli C, Guigni BA, Shulman GI, et al . Impact of CD1d deficiency on metabolism. PLoS One. 2011; 6: e25478.
- Martin-Murphy BV, You Q2, Wang H3, De La Houssaye BA4, Reilly TP5, Friedman JE4, et al . Mice lacking natural killer T cells are more susceptible to metabolic alterations following high fat diet feeding. PLoS One. 2014; 9: e80949.
- Lynch L . Adipose invariant natural killer T cells. Immunology. 2014; 142: 337-346.
- Huh JY, Park YJ2, Ham M, Kim JB . Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol Cells. 2014; 37: 365-371.
- Huh JY, Kim JI, Park YJ, Hwang IJ, Lee YS, Sohn JH, et al . A novel function of adipocytes in lipid antigen presentation to iNKT cells. Mol Cell Biol. 2013; 33: 328-339.
- Strodthoff D, Lundberg AM, Agardh HE, Ketelhuth DF, Paulsson-Berne G, Arner P, et al . Lack of invariant natural killer T cells affects lipid metabolism in adipose tissue of diet-induced obese mice. Arterioscler Thromb Vasc Biol. 2013; 33: 1189-1196.
- Elinav E, Pappo O, Sklair-Levy M, Margalit M, Shibolet O, Gomori M, et al. Adoptive transfer of regulatory NKT lymphocytes ameliorates non-alcoholic steatohepatitis and glucose intolerance in ob/ob mice and is associated with intrahepatic CD8 trapping. J Pathol. 2006; 209: 121-128.
- Margalit M, Shalev Z, Pappo O, Sklair-Levy M, Alper R, Gomori M, et al . Glucocerebroside ameliorates the metabolic syndrome in OB/OB mice. J Pharmacol Exp Ther. 2006; 319: 105-110.
- Schipper HS, Rakhshandehroo M, van de Graaf SF, Venken K, Koppen A, Stienstra R, et al . Natural killer T cells in adipose tissue prevent insulin resistance. J Clin Invest. 2012; 122: 3343-3354.
- Tupin E, Nicoletti A, Elhage R, Rudling M, Ljunggren HG, Hansson GK, et al . CD1d-dependent activation of NKT cells aggravates atherosclerosis. J Exp Med. 2004; 199: 417-422.
- Galkina E, Ley K . Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol. 2009; 27: 165-197.
- Subramanian S, Turner MS, Ding Y, Goodspeed L, Wang S, Buckner JH, et al . Increased levels of invariant natural killer T lymphocytes worsen metabolic abnormalities and atherosclerosis in obese mice. J Lipid Res. 2013; 54: 2831-2841.
- Nakai Y, Iwabuchi K, Fujii S, Ishimori N, Dashtsoodol N, Watano K, et al . Natural killer T cells accelerate atherogenesis in mice. Blood. 2004; 104: 2051-2059.
- Li Z, Lin H, Yang S, Diehl AM . Murine leptin deficiency alters Kupffer cell production of cytokines that regulate the innate immune system. Gastroenterology. 2002; 123: 1304-1310.
- Li Z, Oben JA, Yang S, Lin H, Stafford EA, Soloski MJ, et al . Norepinephrine regulates hepatic innate immune system in leptin-deficient mice with nonalcoholic steatohepatitis. Hepatology. 2004; 40: 434-441.
- Guebre-Xabier M, Yang S, Lin HZ, Schwenk R, Krzych U, Diehl AM . Altered hepatic lymphocyte subpopulations in obesity-related murine fatty livers: potential mechanism for sensitization to liver damage. Hepatology. 2000; 31: 633-640.
- Yang L, Jhaveri R, Huang J, Qi Y, Diehl AM . Endoplasmic reticulum stress, hepatocyte CD1d and NKT cell abnormalities in murine fatty livers. Lab Invest. 2007; 87: 927-937.
- Kremer M, Hines IN, Milton RJ, Wheeler MD . Favored T helper 1 response in a mouse model of hepatosteatosis is associated with enhanced T cell-mediated hepatitis. Hepatology. 2006; 44: 216-227.
- Li Z, Soloski MJ, Diehl AM . Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology. 2005; 42: 880-885.
- Tang T, Sui Y, Lian M, Li Z, Hua J . Pro-inflammatory activated Kupffer cells by lipids induce hepatic NKT cells deficiency through activation-induced cell death. PLoS One. 2013; 8: e81949.
- Kremer M, Thomas E, Milton RJ, Perry AW, van Rooijen N, Wheeler MD, et al . Kupffer cell and interleukin-12-dependent loss of natural killer T cells in hepatosteatosis. Hepatology. 2010; 51: 130-141.
- Ma X, Hua J, Li Z . Probiotics improve high fat diet-induced hepatic steatosis and insulin resistance by increasing hepatic NKT cells. J Hepatol. 2008; 49: 821-830.
- Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, et al . Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology. 2010; 51: 1998-2007.
- Syn WK, Agboola KM, Swiderska M, Michelotti GA, Liaskou E, Pang H, et al . NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut. 2012; 61: 1323-1329.
- Tajiri K, Shimizu Y, Tsuneyama K, Sugiyama T . Role of liver-infiltrating CD3+CD56+ natural killer T cells in the pathogenesis of nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. 2009; 21: 673-680.
- Adler M, Taylor S, Okebugwu K, Yee H, Fielding C, Fielding G, et al . Intrahepatic natural killer T cell populations are increased in human hepatic steatosis. World J Gastroenterol. 2011; 17: 1725-1731.