
Special Article – Vitamin B12
Austin J Nutri Food Sci. 2020; 8(1): 1136.
Conditions and Diseases that Cause Vitamin B12 Deficiency: Form Metabolism to Diseases
Andres E*, Zulfiqar AA and Villalba NL
Department of Internal Medicine, Diabetes and Metabolic Diseases, Hôpitaux Universities de Strasbourg, and Faculty of Medicine, University de Strasbourg, France
*Corresponding author: Andres E, Department of Internal Medicine, Diabetes and Metabolic Diseases, Hôpitaux Universities de Strasbourg, and Faculty of Medicine, University de Strasbourg, Strasbourg, France
Received: January 06, 2020; Accepted: March 02, 2020; Published: March 09, 2020
Introduction
Although vitamin B12 (cobalamin) was isolated almost 60 years ago, its metabolism remains incompletely defined. In practice, cobalamin metabolism is complex and requires many processes and steps, any one of which, if not present, may lead to vitamin B12 deficiency [1,2]. The Figure 1 describes through a synthetic view the different stages of vitamin B12 metabolism used in clinical practice and the corresponding etiologies of cobalamin deficiency [3,4].
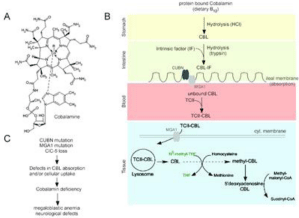
Figure 1: (A); Structure of vitamin B12, (B); Various steps of vitamin B12
(cobalamin [cbl]) metabolism.
For the Practitioner, it should be noted that the clinical manifestations of vitamin B12 deficiency are very numerous, polymorphic, sometimes frustrating, of chronic evolution, or on the contrary severe, of acute revelation (Table 1) [3,4]. The present review summarizes the current knowledge on vitamin B12 metabolism and metabolic pathways in a clinical perspective, with a focus on the etiologies of cobalamin deficiency, especially in adult.
![]()
Hematological manifestations
Neuro–psychiatric manifestations
Digestive manifestations
Other manifestations
Frequent: macrocytosis,
Frequent: polyneuritis
Classic: Hunter’s glossi–
Frequent: Tiredness, loss
Neutrophil hyperseg– mentation , aregenera– tive macrocytary
(especially sensitive),
ataxia, Babinski’s phe– nomenon
tis, jaundice, LDH and bilirubin elevation
of appetite Under study: atrophy of the vaginal mucosa and
(‘‘intramedullary
the vaginal mucosa and
Anemia, medullar mega–
Classic: combined
destruction’’)
chronic vaginal and
Loblastosis (‘‘blue spinal cord’’)
sclerosis of the spinal
Debatable: abdominal
urinary infections (especially mycosis), hypofer– tility and repeated
cord
pain, dyspepsia, nausea,
Rare: isolated thrombo–
Rare: isolated thrombo–
vomiting, diarrhea, dis–
Cytopenia and neutro–
cytopenia and neutro–
turbances in intestinal
miscarriages, venous
Penia, pancytopenia
penia, pancytopenia
functioning
thromboembolic dis–
Very rare: hemolytic anemia, thrombotic microangiopathy (pre– sence of schistocytes)
Under study: changes in the higher functions, dementia, stroke and atherosclerosis (hyper– homocysteinemia), par– kinsonian syndromes, depression, multiple sclerosis
Rare: resistant and recurring mucocuta– neous ulcers
ease, angina (hyperhomocysteinemia)
Table 1: Main clinical features of cobalamin deficiency [2,3,6,14].
Vitamin B12 ingestion and related disorders.
Vitamin B12 sources and dietary recommendations.
Vitamin B12 is produced exclusively by microbial synthesis in the digestive tract of animals. Therefore, animal products are the main sources of cobalamin in the human diet, in particular organ meats (liver, kidney) [2-4]. Other good sources are fish, eggs and dairy products. In foods, hydroxo - methyl- and 5-deoxyadenosylcobalamins are the main cobalamins present. A typical Western diet contributes 3–30 μg of cobalamin per day. The Food and Nutrition Board of the US Institute of Medicine recommends dietary allowance (RAD) of 2.4 μg per day for adults and 2.6 to 2.8 μg per day during pregnancy [5]. The RDA did not distinguish between adults and elderly people though it is questionable whether an intake of 2.4 μg per day can maintain cobalamin status in elderly people with often poor nutrition (malnutrition, in quality and quantity), malabsorption or maldigestion.
Malnutrition, Vegetarianism and Veganism
Vitamin B12 deficiency caused by limited intake of vitamin dietary sources (which requires any animal product intake) is rare, even exceptional, in general population [6,7]. Malnutrition of cobalamin concerns especially vegans and more rarely vegetarians not supplemented by pharmacological vitamin intakes. These nutritional practices, particularly veganism this is currently fashionable in industrialized countries to purge the body and mind, especially among working people in their early 30s and in “up to date” people.
Dietary causes of cobalamin deficiency are common in elderly people who are already malnourished, such as elderly frailty patients in psychiatric hospitals [6] or those living in institutions who may consume inadequate amounts of vitamin B12 containing foods. However, an inadequate intake is not the only explanation for the common cobalamin deficiency in elderly population. Foodcobalamin malabsorption is a significant participating factor in elderly people [6]. Studies focusing on elderly people, particularly those who are in institutions or who are sick and malnourished have suggested a vitamin B12 deficiency prevalence of 30–40% (defined as low vitamin B12 blood level) [6,8]. In this setting, the Framingham study demonstrated a prevalence of 12% among elderly people living in the community [9]. Using stringent definition (presence of clinical and/or biological signs of cobalamin deficiency), we found that vitamin B12 deficiency had a prevalence of 5% in a group of patients (mean age of the patient: 72 years) followed or hospitalized in a tertiary reference hospital in France [3].
In practice, the diagnosis of malnutrition, vegetarianism or veganism is based on patient interview and a comprehensive dietary survey (at least 1 week).
Food-Cobalamin Digestion
Physiology of cobalamin absorption
Dietary cobalamin, which is bound to proteins in food, is released in the acidic environment of the stomach where it is rapidly complexed to the binding protein and transporter haptocorrin (HC), also referred to as the R-binder or trans cobalamin I (Figure 1) [1,10]. About 80% of circulating vitamin B12 are bound to HC and serum cobalamin levels show positive correlation to serum HC concentrations.
Although some unexplained low serum cobalamin concentrations were reported to be caused by mild to severe HC deficiencies, these abnormalities were not accompanied by clinical manifestations of cobalamin deficiency [11,12].
Cobalamin continues its route in the gastrointestinal track and dissociates from HC under the action of pancreatic proteases, followed by its association in the intestine with the intrinsic factor (also known as the S-binder) which is essential for ideal absorption of cobalamin (Figure 1) [1,10]. The intestinal absorption of cobalamin into the enterocytes takes place in the terminal ileum via intrinsic factor receptor cubing. The amount of acid secretion in the gastrointestinal tract plays a critical role in binding of cobalamin to its transporting proteins.
Indeed, homozygous nonsense and missense mutations in the gene encoding the gastric intrinsic factor GIF were reported to cause hereditary juvenile cobalamin deficiency [13]. This metabolism step is also implicated in the physiopathology of the so called Bireme’s disease (see the next section) [1].
Food-cobalamin malabsorption
Food-cobalamin malabsorption (FCM) is a syndrome characterized by the inability of the body to release cobalamin from food or intestinal transport proteins (“maldigestion”), particularly in the presence of hypochlorohydria (Figure 1) [14,15]. Ralph Carmel first characterizes FCM in cases of subtle cobalamin deficiencies [15]. In our experience, this syndrome accounted for 60 to 70% of cases of mild to severe vitamin B12 deficiency in elderly patients [16]. The principal characteristics of this syndrome are listed in Table 2. FCM is caused primarily by atrophic gastritis [14]. Other factors that contribute to FCM are: chronic infection with Helicobacter pylori and intestinal microbial proliferation, situations in which cobalamin deficiency can be corrected by antibiotic treatment [14,17]; longterm ingestion of anti-acid agents such as H2-receptor antagonists and proton-pump inhibitors (e.g., omeprazole, pantoprazole, esomeprazole, etc.), particularly among patients with Zollinger- Ellison syndrome (gastronome) [18], and long term intake of biguanides (metformin) [19,20]. In addition, other FCM inducers include: chronic alcoholism, especially in malnourished patients; surgery or gastric reconstruction (e.g., bypass surgery for obesity, partial gastrectomy for benign gastric tumor or malignant gastric tumors, etc.); partial exocrine pancreatic failure (e.g., chronic alcohol intake, cystic fibrosis, etc.); and rarely Soren’s syndrome and systemic sclerosis or HIV [14].
![]()
Criteria for food-cobalamin malabsorption
Low serum cobalamin (vitamin B12) levels
Normal results of Schilling test using free cyanocobalamin labeled with cobalt-58 or abnormal results of derived Schilling test ‡
No anti-intrinsic factor antibodies
No dietary cobalamin deficiency
Presence of an associated conditions or agents (e.g., atrophic gastritis, Helicobacter pylori infection, partial gastrectomy, gastric by-pass, pancreatic insufficiency (alcohol abuse), intestinal bacterial overgrowth, long term anti-acid agents (proton-pump inhibitors) or biguanides (metformin) intake
‡ Derived Schilling tests use food-bound cobalamin (e.g., egg yolk, chicken and fish proteins)
Table 2: Food-cobalamin malabsorption syndrome [14].
It is to note that in case of FCM, patients can absorb “unbound” cobalamin through intrinsic factor or passive diffusion mechanisms [10,14]. Thus the recognition of the syndrome permits new developments of oral cobalamin therapy using free crystalized cobalamin that is readily absorbed [21]. In practice, the diagnosis of FCM is to date based on the exclusion of the main others causes of vitamin B12 deficiency, in connection with the fact that the Schilling’s test is no longer currently available [4]. Health care providers need to be aware of FCM, since it is easy to treat and treatment can prevent serious late consequences of vitamin B12 deficiency.
Vitamin B12 Absorption
Physiology
Absorption depends mainly on intrinsic factor (IF), which is secreted by the gastric mucosa. IF binds cobalamin forming a complex that is absorbed by the terminal ileum (Figure 1) [1,10]. This mechanism is responsible for at least 60% absorption on oral cobalamin [1]. This complex is located at the apical side of brushborder membranes (BBMs) of polarized epithelia, such as the intestinal apical BBM.
It consists of the intrinsic factor-vitamin B12 receptor named cubing, a 460 kDa peripheral membrane glycoprotein, encoded by the CUBN gene which was mapped to chromosomal region 10p12.33-p13 [22], and the 48 kDa amnion less (AMN) protein encoded by the AMN gene, a gene localized on human chromosome 14 [23]. In this setting, the human mega in /gp330/LRP-2 receptor, encoded by the LRP-2 gene located on chromosome 2q24-q31 [24], may play an important role in the stability of the cubing /AMN complex [10,25- 29]. It is noteworthy that the interaction of these factors is Ca+2 dependent [26-29].
Cubing and megalin are also expressed in the apical side of proximal kidney tube and are considered responsible for cobalamin reuptake into the circulation [10].
Biermer’s or addison’s disease
In adults, vitamin B12 deficiency is classically caused by Biermer’s also named Addison’s disease, formerly known as “pernicious anemia” [30,31]. This disorder is an auto-immune disease characterized by: the destruction of the gastric mucosa, especially fundal, associated with a primarily cell-mediated auto-immune process; and the presence of various antibodies, especially anti-intrinsic factor antibodies and gastric parietal anti-cell antibodies that target the H+/K+ ATPase a and β subunits [30,31]. Pernicious anemia has a genetic component [30].
In this context, pernicious anemia is associated with other immunologic diseases such as Soren’s syndrome, Hashimoto’s disease, type 1 diabetes mellitus, and celiac disease [30,32]. In our experience, Biermer’s disease accounted for 30 to 40% of cases of cobalamin deficiency in adults, and more than 60% in severe vitamin B12 deficiencies [4]. In this later situation, hematological (e.g. macrocytic anemia) or psycho-neurological manifestations (e.g. medullar combined sclerosis) are commonly observed [32].
In practice, the diagnosis of Biermer’s or Addison’s disease is based on the presence of intrinsic factor antibodies in serum (specificity › 98% and sensitivity around 50%) or biopsy-proven autoimmune atrophic gastritis (Table 3) [1,30]. The presence of Helicobacter pylori infection in gastric biopsies is an exclusion factor.
![]()
Criteria for Biermer’s disease (pernicious anemia)
Low serum cobalamin (Vitamin B12) levels
Abnormal results of Schilling test using free cyanocobalamin labeled with cobalt 58 ‡
Presence of anti-intrinsic factor antibodies (sensibility of 50%, specificity >98%)
Presence of an auto-immune gastritis (especially fundal), with absence of Helicobacter pylori in the status phase of the disease
Associated conditions: auto-immune disorders (e.g., Sjögren’s syndrome, Hashimoto’s disease, type 1 diabetes mellitus, and celiac disease)
Predisposition of gastric cancer
‡Schilling test is not used anymore in clinical practice.
Table 3: Biermer’s or Addison’s disease [1,30,32].
It is important to note that of Biermer’s disease require a long term gastric follow-up (upper-endoscopy with biopsies, every year in case of gastric lesions or every 2 to 5 years in the absence of detectable lesion), because this disorder favors the emergence of various cancers of the stomach [1,4].
Cobalamin malabsorption
Since the 1980s, the prevalence of vitamin B12 malabsorption declined, owing mainly to the decreasing frequency of gastrectomy (due to the provision of antacid drugs) and terminal small intestine surgical resection [4,10].
However, several disorders commonly seen in gastroenterology practice might, to date, be associated with cobalamin malabsorption [10]. These disorders include: exocrine pancreas’ function deficiency following chronic pancreatitis (usually alcoholic), lymphomas or tuberculosis (of the intestine), celiac disease, Crohn’s disease, Whipple’s disease, and uncommon celiac disease [4].
In practice, the diagnosis is based on patient interview (personal surgical history), a full clinical examination and digestive explorations in doubt.
Genetic Disorders of Cobalamin Malabsorption
In this setting, the Imerslund-Gräsbeck syndrome or megaloblastic anemia due to selective cobalamin malabsorption with proteinuria is a vitamin B12 deficiency leading to megaloblastic anemia in childhood and is corrected by parenteral administration of vitamin B12 [10,34- 38]. This pathology is mainly localized in Finland, Norway and the Mediterranean Muslim countries. The disease was described around 1960 simultaneously by Olga Imerslund, a Norwegian pediatrician and Ralph Gräsbeck, a Finnish physician-biochemist [33]. Other manifestations of the disease include stature-weight loss, frequent infections and neurological signs. Proteinuria without kidney damage is present in half of the patients. Abnormalities of the urinary tract are sometimes observed.
Mutations in CUBN were reported to cause hereditary megaloblastic anemia 1 (MGA1), a rare autosomal recessive disorder affecting human subjects with neurological symptoms and juvenile megaloblastic anemia [34-36]. Two principal mutations were identified in Finnish patients (FM), a 3916C→T missense mutation named FM1 changing a highly conserved proline to leucine (P1297L) in CUB domain 8, suggesting that this proline is functionally crucial in cubilin, and one point mutation (FM2) in the intron interrupting CUB domain 6 responsible for in-frame insertions producing truncated cubilin [10,35]. Other mutations were also uncovered but were subsequently identified as polymorphisms after their detection in normal individuals in the general population. The cubilin P1297L mutation associated with hereditary MGA1 was reported to cause impaired recognition of the cobalamin-IF complex by cubilin [36]. Moreover, mutation in AMN was reported in recessive hereditary MGA1 [10,36] and hence was demonstrated to be crucial for a functional cobalamin-IF receptor [10]. This study demonstrated that homozygous mutations affecting exons 1-4 of the human AMN gene translated into selective malabsorption of vitamin B12, a phenotype associated with hereditary MGA1. Another study reported AMN deletion mutants in dogs with selective intestinal malabsorption of cobalamin associated with urinary loss of low molecular weight protein reminiscent of the human Imerslund-Gräsbeck syndrome (IGS a.k.a. MGA1) [34,35]. The authors showed that these mutations in the AMN gene abrogated AMN expression and blocked cubilin processing and targeting to the apical membrane [37]. The essential AMN-cubilin interaction was recapitulated and validated in a heterologous cell-transfection model, hence explaining the molecular basis of intestinal cobalamin malabsorption syndrome [10,38].
Of particular interest for the practitioner in this section of malabsorption is the observation that about 1 to 5% of free vitamin B12 (or crystalline cobalamin) is absorbed along the entire intestine by passive diffusion [10]. This absorption explains the mechanism underlying oral cobalamin treatment of vitamin B12 deficiencies [39]. Our working group has developed an effective oral treatment for FCM [40] and pernicious anemia [41] using crystalline cobalamin (cyanocobalamin) (Table 5). Oral cobalamin has been proposed as a way of avoiding the discomfort, inconvenience and cost of monthly injections.
Cobalamin Transport to Blood and Tissues
Physiology
After vitamin B12 is absorbed at the BBM-blood barrier, it dissociates from the intrinsic factor and reaches the systemic circulation where it associates with Trans cobalamin II (TCII) (Figure 1) [1,10,38]. The kidney represents an essential organ were body vitamin B12 stores are maintained and studies demonstrated that the kidney regulates plasma vitamin B12 levels by maintaining a pool of unbound cobalamin that can be released in case of vitamin B12 deficiency [10,42]. The insular cobalamin-TCII complex uptake is achieved through megalin (LRP2), and trans cobalamin II receptor (TCII-R)-mediated endocytosis which plays a crucial role in cobalamin homeostasis [32,43].
Following cobalamin-TCII cellular uptake, TCII undergoes lysosomal digestion, which allows cobalamin separation from TCII and its cytoplasmic transfer. It has been estimated that there is a delay ranging from 5 and 10 years between the onset of cobalamin deficiency and the appearance of clinical manifestations, due to largehepatic stores (› 1.5 mg) and the enterohepatic cycle ensuring re-absorption of the vitamin in the gastrointestinal tract [1,10]. Also the reabsorption of TCII-bound cobalamin in the proximal tubules limits the loss of B12 in urine.
The average vitamin B12 content is approximately 1.0 mg in healthy adults, with 20-30 μg found in the kidneys, heart, spleen and brain. Estimates of total vitamin B12 body content for adults range from 0.6 to 3.9 mg with mean values of 2-3 mg. The normal range of vitamin B12 plasma concentrations is 150-750 pg/ml, with peak levels achieved 8-12 hours after ingestion of a single dose of the vitamin.
Part of the cobalamin serves as a cofactor for methionine synthase-mediated homocysteine catabolism into methionine and methyl tetrahydrofolate reductase (MTHFR)-mediated formation of the vitamin B9 biologically active form, tetrahydrofolate, which is then involved in the synthesis of purines and pyrimidines (Figure 1) [10,44]. In clinical practice, several of these molecules are implicated in the definitions of vitamin B12 deficiency (Table 4). The other part of vitamin B12 is transferred to the mitochondria where it is transformed into adenosyl-B12, an important cofactor in methylmalonyl-coenzyme a mutase-mediated formation of succinyl- CoA from methylmalonyl-CoA, the product of odd-chain fatty acid and some amino acid catabolism. Hence, cobalamin deficiency will cause homocysteine accumulation, increased methylmalonyl-CoA levels and decreased MTHFR activity. These changes are translated into several abnormalities including folate deficiency and subsequent inhibition of purines and pyrimidines formation essential for RNA and DNA synthesis [10,38].
![]()
Serum cobalamin levels <150 pmol/l and clinical features and/or hematological anomalies related to cobalamin deficiency.
Serum cobalamin levels <150 pmol/l (<200 pg/ml) on two separate occasions.
Serum cobalamin levels <150 pmol/l and total serum homocysteine levels >13 mmol/l or methyl–malonic acid levels >0.4 mmol/l (in the absence of kidney failure or atheroma and of Methylene Tetra Hydro Folate Reductase (MTHFR) deficiency and in the absence of folate and vitamin B6 deficiencies).
Low serum holotranscobalamin levels <35 pmol/l.
Table 4: Definitions of cobalamin (Vitamin B12) deficiency [3,4,6,10,14].
![]()
Pernicious anaemia
Intake deficiency and food-cobalamin malabsorption
Parenteral administration (intramuscular)
Cyanocobalamin:
Cyanocobalamin:
1,000 μg per day for 1 week
than 1,000 μg per week for 1 month
than 1,000 μg per each month, for life
(1,000 to 2,000 μg per day for at least 1 to 3 months in case of severe neurological manifestations)
1,000 μg per day for 1 week
than 1,000 μg per week for 1 month
than 1,000 μg per each 1 or 3 months, until the cobalamin deficiency cause is corrected
(1,000 μg per day for at least 1 to 3 months in case of severe neurological manifestations)
Oral administration
Cyanocobalamin:
Cyanocobalamin:
1,000 μg per day for life*
1,000 μg per day for 1 month than 125 to 1,000 μg per day, until the cobalamin deficiency cause is corrected*
*The effect of oral cobalamin treatment in patients presenting with severe neurological manifestations has not yet been adequately documented.
Table 5: Recommendations for oral vitamin B12 treatment [39].
Genetic Disorders of Cobalamin Transport
It is worth mentioning that TCII is responsible for the cellular uptake of B12 in most tissues and that TC deficiency is associated with severe megaloblastic anemia [45-48] and developmental disorders (see the review in [48]).
Impaired megalin function has not been associated with cobalamin deficiency so far; however inappropriate megalin signaling has been shown to cause deleterious effects as a consequence of cobalamin uptake inhibition in tissues. This was particularly the case where mutations in the human LRP2 gene encoding megalin were recently described to cause Donna -Barrow and facio-oculo-acoustico-renal syndromes. Patients affected with these rare autosomal recessive disorders display severe malformations with proteinuria [47].
The clinical manifestations of the metabolic abnormalities are hereditary megaloblastic anemia, neurological defects, malformations, increased cardiovascular thrombotic risk and renal disease, and methyl malonic acidemia [10,38,44]. Functional cobalamin deficiency can also be caused by defects in the intracellular processing of cobalamin such as abnormal lysosomal digestion of the TCII-cobalamin complex, and subsequent defective lysosomal release of cobalamin, and abnormalities in intracytoplasmic cobalamin metabolism with all the consequences on biochemical reactions in which cobalamin acts as an important cofactor [10,44].
Conclusion
In this paper, we presented the main etiologies of vitamin B12 deficiency in relation to different steps of the cobalamin transport and metabolism. However to date, many causes of cobalamin deficiency remained unknown. These causes include mutations in genes encoding important proteins of the cobalamin transport or metabolic pathway. Moreover, many clinically diagnosed vitamin B12 deficiency remain unexplained and molecular tools aimed at targeting genes involved in vitamin B12 absorption and cellular uptake signaling pathways will pave the way for new therapeutic approaches to efficiently treat functional cobalamin deficiency.
Dietary vitamin B12 is absorbed in food bound to proteins and undergo acidic gastric digestion. The released cbl is attached to the R-binder haptocorrin and transported to the intestine following pancreatic proteases processing. The unbound cbl is then associated in the gut with the gastric-produced intrinsic factor (IF). This association is necessary for intestinal cbl absorption through a complex of endocytic receptors and proteins, including the endocytic receptor cubilin (CUBN) and the apical membrane protein amnion less (AMN), which is stabilized by two other proteins, namely the receptor megalin/LRP-2 and its binding protein receptor associated protein (RAP) which also binds to cubilin. After it absorption free cobalamin reaches the systemic circulation where it associates with Tran’s cobalamin II (TCII) and subsequently the cbl-TCII complex is up taken in cells through it binding to megalin/LRP-2 and TCII receptor (TCII-R). Intracellularly, the complex is dissociated following lysosomal digestion. Part of cbl serves as a cofactor for the methionine synthase (MS) mediated transformation of homocysteine into methionine and for methyl-tetrahydrofolate reductase-mediated formation of tetrahydrofolate (THF) a precursor of purine and pyrimidine necessary for nucleic acid synthesis. The other fraction of cbl reaches the mitochondria where it forms adenosyl-cbl, a cofactor for the methyl-malonyl mutase-mediated catabolism of methylmalonyl coA. (C): Mutations in genes encoding the intrinsic factor, cubilin, amnion less or Trans cobalamin II or its receptor provoke defects in cbl absorption and/or cellular uptake which translates into functional cbl deficiency and its clinical manifestations.
Acknowledgment
We especially want to thank Dr. Nassim Dali-Youcef who contributed to this chapter through his expertise and assistance to our works on the management of vitamin B12 deficiency in adults.
References
- Nicolas JP, Gueant JL. Absorption, distribution and excretion of vitamin B12. Ann Gastroenterol Hepatol (Paris). 1994; 30: 270-276.
- Markle HV. Cobalamin. Crit Rev Clin Lab Sci. 1996; 33: 247-356.
- Andrès E, Loukili NH, Noel E, Kaltenbach G, Abdelgheni MB, Perrin AE, et al. Vitamin B12 (cobalamin) deficiency in elderly patients. CMAJ. 2004; 171: 251-254.
- Stabler SP. Clinical practice. Vitamin B12 deficiency. N Engl J Med. 2013; 368: 149-160.
- Medicine I. Dietary reference intake of thiamin, riboflavin, niacin, vitamin B6, folate, vitamin B12, panthotenic acid, biotin and choline Food and Nutrition Board, Washington DC National Academies Press. 1998.
- Matthews JH. Cobalamin and folate deficiency in the elderly. Baillieres Clin Haematol. 1995; 8: 679-697.
- Pawlak R, Lester SE, Babatunde T. The prevalence of cobalamin deficiency among vegetarians assessed by serum vitamin B12: a review of literature. Eur J Clin Nutr. 2014; 68: 541-548.
- van Asselt DZ, Blom HJ, Zuiderent R, Wevers RA, Jakobs C, van den Broek WJ, et al. Clinical significance of low cobalamin levels in older hospital patients. Neth J Med. 2000; 57: 41-49.
- Lindenbaum J, Rosenberg IH, Wilson PW, Stabler SP, Allen RH. Prevalence of cobalamin deficiency in the Framingham elderly population. Am J Clin Nutr. 1994; 60: 2-11.
- Dali-Youcef N, Andrès E. An update on cobalamin deficiency in adults. QJM. 2009; 102: 17-28.
- Carmel R. R-binder deficiency. A clinically benign cause of cobalamin pseudo deficiency. JAMA. 1983; 250: 1886-1890.
- Carmel R. Mild transcobalamin I (haptocorrin) deficiency and low serum cobalamin concentrations. Clin Chem. 2003; 49: 1367-1374.
- Tanner SM, Li Z, Perko JD, Oner C, Cetin M, Altay C, et al. Hereditary juvenile cobalamin deficiency caused by mutations in the intrinsic factor gene. Proc Natl Acad Sci USA. 2005; 102: 4130-4133.
- Andrès E, Affenberger S, Vinzio S, Kurtz JE, Noel E, Kaltenbach G, et al. Food-cobalamin malabsorption in elderly patients: clinical manifestations and treatment. Am J Med. 2005; 118: 1154-1159.
- Carmel R. Malabsorption of food cobalamin. Baillieres Clin Haematol. 1995; 8: 639-655.
- Andrès E, Goichot B, Schlienger JL. Food-cobalamin malabsorption: a usual cause of vitamin B12 deficiency. Arch Intern Med. 2000; 160: 2061-2062.
- Kaptan K, Beyan C, Ural AU, Cetin T, Avcu F, Gulsen M, et al. Helicobacter pylori--is it a novel causative agent in Vitamin B12 deficiency? Arch Intern Med. 2000; 160: 1349-1353.
- Jung SB, Nagaraja V, Kapur A, Eslick GD. Association between vitamin B12 deficiency and long-term use of acid-lowering agents: a systematic review and meta-analysis. Intern Med J. 2015; 45: 409-416.
- Bauman WA, Shaw S, Jayatilleke E, Spungen AM, Herbert V. Increased intake of calcium reverses vitamin B12 malabsorption induced by metformin. Diabetes Care. 2000; 23: 1227-1231.
- Andrès E, Noel E, Goichot B. Metformin-associated vitamin B12 deficiency. Arch Intern Med. 2002; 162: 2251-2252.
- Andrès E, Vogel T, Federici L, Zimmer J, Kaltenbach G. Update on oral cyanocobalamin (vitamin B12) treatment in elderly patients. Drugs Aging. 2008; 25: 927-932.
- Kozyraki R, Kristiansen M, Silahtaroglu A, Hansen C, Jacobsen C, Tommerup N, et al. The human intrinsic factor-vitamin B12 receptor, cubilin: molecular characterization and chromosomal mapping of the gene to 10p within the autosomal recessive megaloblastic anemia (MGA1) region. Blood. 1998; 91: 3593-600.
- Fyfe JC, Madsen M, Hojrup P, Christensen EI, Tanner SM, de la Chapelle A, et al. The functional cobalamin (vitamin B12)-intrinsic factor receptor is a novel complex of cubilin and amnionless. Blood. 2004; 103: 1573-1579.
- Saito A, Pietromonaco S, Loo AK, Farquhar MG. Complete cloning and sequencing of rat gp330/”megalin,” a distinctive member of the low density lipoprotein receptor gene family. Proc Natl Acad Sci USA. 1994; 91: 9725- 9729.
- Ahuja R, Yammani R, Bauer JA, Kalra S, Seetharam S, Seetharam B. Interactions of cubilin with megalin and the product of the amnionless gene (AMN): effect on its stability. Biochem J. 2008; 410: 301-308.
- Moestrup SK, Birn H, Fischer PB, Petersen CM, Verroust PJ, Sim RB, et al. Megalin-mediated endocytosis of transcobalamin-vitamin-B12 complexes suggests a role of the receptor in vitamin-B12 homeostasis. Proc Natl Acad Sci USA. 1996; 93: 8612-8617.
- Birn H, Verroust PJ, Nexo E, Hager H, Jacobsen C, Christensen EI, et al. Characterization of an epithelial approximately 460-kDa protein that facilitates endocytosis of intrinsic factor-vitamin B12 and binds receptor-associated protein. J Biol Chem. 1997; 272: 26497-26504.
- Kristiansen M, Kozyraki R, Jacobsen C, Nexo E, Verroust PJ, Moestrup SK. Molecular dissection of the intrinsic factor-vitamin B12 receptor, cubilin, discloses regions important for membrane association and ligand binding. J Biol Chem. 1999; 274: 20540-20544.
- Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol. 2002; 3: 256-266.
- Rojas Hernandez CM, Oo TH. Advances in mechanisms, diagnosis, and treatment of pernicious anemia. Discov Med. 2015; 19: 159-168.
- Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med. 1997; 337: 1441-1448.
- Andrès E, Affenberger S, Zimmer J, Vinzio S, Grosu D, Pistol G, et al. Current hematological findings in cobalamin deficiency. A study of 201 consecutive patients with documented cobalamin deficiency. Clin Lab Haematol. 2006; 28: 50-56.
- Imerslund O. Idiopathic chronic megaloblastic anemia in children. Acta Paediatr. 1960; 49: 1-115.
- Grasbeck R. Familiar selective vitamin B12 malabsorption with proteinuria. A pernicious anemia-like syndrome. Nord Med. 1960; 63: 322-323.
- Aminoff M, Carter JE, Chadwick RB, Johnson C, Grasbeck R, Abdelaal MA, et al. Mutations in CUBN, encoding the intrinsic factor-vitamin B12 receptor, cubilin, cause hereditary megaloblastic anaemia 1. Nat Genet. 1999; 21: 309- 313.
- Kristiansen M, Aminoff M, Jacobsen C, de La Chapelle A, Krahe R, Verroust PJ, et al. Cubilin P1297L mutation associated with hereditary megaloblastic anemia 1 causes impaired recognition of intrinsic factor-vitamin B(12) by cubilin. Blood. 2000; 96: 405-409.
- He Q, Madsen M, Kilkenney A, Gregory B, Christensen EI, Vorum H, et al. Amnionless function is required for cubilin brush-border expression and intrinsic factor-cobalamin (vitamin B12) absorption in vivo. Blood. 2005; 106: 1447-1453.
- Quadros EV. Advances in the understanding of cobalamin assimilation and metabolism. Br J Haematol. 2010; 148: 195-204.
- Andrès E, Fothergill H, Mecili M. Efficacy of oral cobalamin (vitamin B12) therapy. Expert Opinion Pharmacotherapy. 2010; 11: 249-256.
- Andrès E, Kurtz JE, Perrin AE, Maloisel F, Demangeat C, Goichot B, et al. Oral cobalamin therapy for the treatment of patients with food-cobalamin malabsorption. Am J Med. 2001; 111: 126-129.
- Andrès E, Henoun Loukili N, Noel E, Maloisel F, Vinzio S, Kaltenbach G, et al. Oral cobalamin (daily dose of 1000 μg) therapy for the treatment of patients with pernicious anemia. An open label study of 10 patients. Curr Ther Research. 2005; 66: 13-22.
- Birn H. The kidney in vitamin B12 and folate homeostasis: characterization of receptors for tubular uptake of vitamins and carrier proteins. Am J Physiol Renal Physiol. 2006; 291: F22-36.
- Yammani RR, Seetharam S, Dahms NM, Seetharam B. Transcobalamin II receptor interacts with megalin in the renal apical brush border membrane. J Membr Biol. 2003; 193: 57-66.
- Fowler B. Genetic defects of folate and cobalamin metabolism. Eur J Pediatr. 1998; 157: S60-66.
- Li N, Rosenblatt DS, Kamen BA, Seetharam S, Seetharam B. Identification of two mutant alleles of transcobalamin II in an affected family. Hum Mol Genet. 1994; 3: 1835-1840.
- Teplitsky V, Huminer D, Zoldan J, Pitlik S, Shohat M, Mittelman M. Hereditary partial transcobalamin II deficiency with neurologic, mental and hematologic abnormalities in children and adults. Isr Med Assoc J. 2003; 5: 868-872.
- Kantarci S, Al-Gazali L, Hill RS, Donnai D, Black GC, Bieth E, et al. Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai- Barrow and facio-oculo-acoustico-renal syndromes. Nat Genet. 2007; 39: 957-959.
- Green R, Allen LH, Bjørke-Monsen AL, Brito A, Guéant JL, Miller JW, Molloy AM, Nexo E, Stabler S, Toh BH, Ueland PM, Yajnik C. Vitamin B12 deficiency. Nat Rev Dis Primers. 2017; 3: 17040.