
Case Report
Austin J Nutr Metab. 2017; 4(2): 1048.
Dilated Cardiomyopathy due to Hypocalcemia Secondary to Congenital Hypoparathyroidism in a Newborn with DiGeorge Syndrome
Al Mutair Angham1,2* and Hanan Al Azkawi1
¹Department of Pediatrics, King Abdullah Specialized Children’s Hospital (KASCH), Saudi Arabia
²King Saud bin Abdulaziz University for Health Sciences, College of Medicine, Saudi Arabia
*Corresponding author: Al Mutair Angham, Department of Pediatrics, Endocrine Division, King Abdullah Specialized Children’s Hospital (KASCH), Saudi Arabia
Received: May 09, 2017; Accepted: May 30, 2017; Published: June 21, 2017
Abstract
Hypocalcemia is a rare but reversible cause of dilated cardiomyopathy (DCM) with limited cases being reported in the literature. Vitamin D deficiency is main cause of hypocalcemia in almost all reported cases. We report a case of DiGeorge syndrome with primary hypoparathyroidism presented with hypocalcemic dilated cardiomyopathy. After calcium and vitamin D replacement therapy, the patient showed a rapid recovery of the cardiac function.
Keywords: Vitamin D deficiency; Dilated cardiomyopathy; DiGeorge syndrome
Introduction
DCM has an estimated incidence of 1.13 cases per 100,000 children. Diagnosing the primary etiology occurs in fewer than half of these children but significantly improves their outcome [1]. Dilated cardiomypathy mostly idiopathic, however, infection and metabolic causes has been identified in some cases in which the defect in myocardial contractility is irreversible [2].
Calcium has a central role in myocardial contraction coupling, and hypocalcemia reduces myocardial function. Congestive cardiac failure (CCF) due to hypocalcemia is also reported, though rare [3]. The incidence of congestive heart failure due to hypocalcemia is quite rare in clinical practice. Several cases of hypocalcemic cardiomyopathy have been reported [4].
Hypocalcemia causing DCM is reversible with complete recovery after normalization of serum calcium. In isolated case reports, a rare cause of dilated cardiomyopathy (DCM) has been nutritional hypocalcemic rickets [1]. Hypoparathyroidism as a cause of hypocalcemia is well known in many diseases and syndromes. However, hypocalcemic DCM secondary to hypoparathyroidism has been reported in adult [5,6]. We report a first case of a newborn confirmed DiGeorge syndrome presented with hypocalcemic dilated cardiomyopathy due to hypoparathyroidism.
Case Presentation
FA is now 6 years old boy who was full term, normal delivery with a birth weight of 2.8 kg. Discharged home in 2nd day of life with no medical problem. He presented at age of 3 weeks with poor/ interrupted feeding, shortness of breath, tachycardia and vomiting for 3 days. On examination, he was tachycardiac, Pulse rate 160/ min, Respiratory Rate (RR) =40 frequency/minute, Blood Pressure 84/58 mmHg, cold extremities, hypoexic with oxygen saturation of 89% on room air corrected to 100% with oxygen supplement. Weight, length and head circumference were on 50%. He was noticed to have dysmorphic features in form of: micrognathia, low-set small posteriori rotated ears, hypertelorism, down slanting short palpebral fissures, narrow alae nasi with broad base of the nose and short philturm (Figure 1).
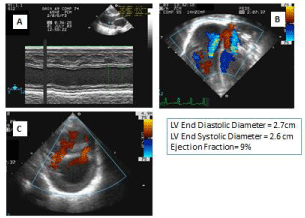
Figure 1: First ECHO.
A: Left ventricle M-mode echocardiography with poor contractility.
B: Moderate tricupid and mitral valves regurgitation by echocardiography.
C: Two muscular ventricular septal defects by echocardiography.
Cardiovascular examination revealed 1st and 2nd heart sound were normal, a grade 2/6 ejection systolic murmur and loud 3rd heart sound with a gallop rhythm (heard all over pericardium) more in the left lower sternal border. Apex is at 6th intercostal space, midclavicular line with positive right ventricle heaves. All pulses were felt with no radio femoral delay with perfusion delay of 6 seconds in his extremities. On the chest exam there were bilateral basal crackles. Abdominal examination revealed hepatomegaly with liver span 9 centimeter. CNS examination was unremarkable.
Chest x-ray film showed significant cardiomegaly (80% of Cardiothoracic Ratio) and congested pulmonary vessels. Electrocardiography (ECG) showed prolonged QT interval.
Echocardiogram (Echo) revealed Left Ventricle M-Mode Echocardiography with poor contractility with an ejection fraction of 9%, Moderate Tricuspid and Mitral Valves Regurgitation by Echo and Two muscular Ventricular Septal Defects (Figure 2).
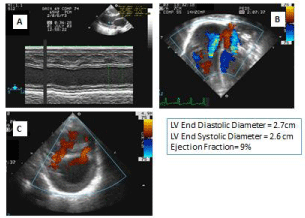
Figure 2: Follow up ECHO at 3 weeks of age and at 6 years of age.
A: Left ventricle M-Mode echocardiography with good contractility.
B: Mild tricuspid and mitral valves regurgitation by echocardiography.
C: One small muscular ventricular septal defect left on last echocardiography.
Baby was admitted to Cardiac Ward and started on antifailure medications (diuretics and captopril). The baby had increased respiratory distress with desaturation and required assisted mechanical ventilation in intensive care unit. He also required inotropic support with dobutamin infusion. During the same time seizures episodes was noticed, which controlled with midazolam.
Blood sample was send for some investigations and surprisingly showed (method used AEROSET and ARCHITECT c8000 systems) very low total Calcium (Total Ca): 0.99 mmol/L (2.25-2.75 mmol/L), very low ionized Calcium (Ca+): 0.38 mmol/L (1.16-1.32 mmol/L), high Phosphorous (PO4): 1.86 mmol/L (1.45-2.16 mmol/L), Magnesium (Mg) 0.60 mmol/L (.65-1.05 mmol/L) (Table 1). Parathyroid hormone was within normal range: 22.0 pg/ml (12- 72 pg/ml), but it is inappropriately low for the degree hypocalcaemia (ARCHITECT system). Vitamin D level was within the normal limit.
![]()
At presentation
4 days later
8 days later
At 2 years
Total calcium (2.20-2.70mmol/L)
0.99
1.49
2.06
2.1
Ionized calcium (1.16-1.32mmol/L)
0.38
0.75
1.18
1.35
Phosphorus (0.74-1.52mmol/L)
1.86
2.20
1.80
1.90
Magnesium (0.71-0.95mmol/L)
0.60
0.75
0.71
0.86
Table 1: Bone profile at presentation and on follow up.
FA has dysmorphic features as described in the examination so he had chromosomal analysis. The chromosomal analysis revealed chromosomal count of 46 XY; a small interstitial deletion was detected in the pericentric region of the long arm of a chromosome 22 homology. The FISH test (Fluorescence In Situ Hybridization) showed a deletion in the critical region of DiGeorge syndrome on Ch 22q11. Parent’s study was negative Ch 22q11 microdeletion.
So the baby managed with intravenous calcium gluconate infusion as well as oral calcium and vitamin D supplements. He improved clinically as his calcium normalizes. He was able to weaned off ventilation and no more seizure was observed. The cardiac function also improved, the inotropes were gradually tapered and then discontinued and antifailure medications were stopped three months later.
Repeated Echo 1 week after his presentation showed improvement in ejection fraction up to 60% and after 3 months the Echo showed normal left ventricular function, Left Ventricle M-Mode Echocardiography with good contractility, Mild Tricuspid and Mitral Valves Regurgitation by Echocardiography, One small muscular Ventricular Septal Defect left on last echocardiography.
FA is now 6 years old on regular follow up. He is still on alfacidol 0.6 microgram per day. He has delayed motor and cognitive development. The calcium level remained above 2.00 mmol/L (2.20-2.70 mmol/L), PO4 between 1.4-1.9 mmol/L (0.74-1.52 mmol/L) and Mg between 0.73-0.91 mmol/L (0.71-0.95 mmol/L). The echocardiogram repeated after one and half year again showed normal left ventricular function.
Other investigations done looking for other causes of dilated cardiomyopathy were negative. These are metabolic screen (ammonia, lactate, carnitine level) and viral serology (Adenovirus and enterovirus serology).
Discussion
Ionized calcium has a central role for regulating myocardial contraction. During the cardiac action potential is activated, ionized calcium (Ca+) enter intracellular through depolarization activated calcium channels. Entered ionized calcium triggers calcium release from the sarcoplasmic reticulum (SR). Ca2+ bind to the myofilaments proteins such as troponin C initiate contraction of myocardium [7]. There are two main ways to change contractility of myocardium. One is altering of amplitude or duration of Ca2+ transient; another is altering of sensitivity of the myofilaments to Ca2+ [7]. Therefore, hypocalcemia induced DCMP developed by the altering of amplitude or duration of Ca2+ transient [8].
The most common cause of hypocalcemia in pediatric age group is nutrional rickets mostly due to vitamin D deficiency [9-11]. In one study a hospital database search was conducted from year 1997 to year 2007 to identify patients with confirmed vitamin D deficiency in addition to DCM. Four exclusively breast-fed African American infants were identified. These infants presented with congestive heart failure secondary to DCM and, at their admission, were found to have laboratory evidence consistent with hypocalcemic rickets. These patients responded dramatically to treatment with vitamin D and calcium, and cardiac function returned to normal within months [9].
In another series of 15 Indian infants (aged between 45 days to 5 months) presented with severe left ventricular dysfunction, were found to have hypocalcemia with or without hypomagnesemia. Vitamin D deficiency was identified as the main cause of hypocalcemia. These children improved on supplementation of vitamin D and calcium [12].
Parathyroid hormone has been known to stimulate renal calcium ion re-absorption [13,14]. In the parathyroidectomy rats, sodium and calcium ion exchange activity was decreased by 40% and this activity was restored by infusion of PTH [10]. Reduced urinary excretion of sodium leads water retention and it may cause heart failure [10,15].
Hypocalcemia due to hypoparathyroidism is associated with reversible cadiomyopathy and has been infrequently reported in the literature, most commonly with idiopathic hypoparathyroidism in adults [5,6,16]. However there are no reported cases in pediatric.
FA is first case reported with congenital hypoparathyroidism in a child with confirmed DiGeorge syndrome presented with hypocalcemic DCM. In our case he presented in very young age, 3 weeks of age, with severe hypocalcemia induced dilated cardiomyopathy (DCM) and inappropriately low parathyroid hormone. His vitamin D level was measured and it was normal. FISH analysis was requested based on his dysmorohic features and clinical presentation of hypoparathydroidism induced hypocalcemia. It confirms the diagnosis of DiGeorge syndrome. This syndrome is characterized by genetic deletions on the long arm of one of the two 22nd chromosomes (22q11.2 deletion syndrome) [17,18]. This deletion cause defect in development of the third and fourth branchial pouches (pharyngeal pouches). Resulting in the clinical features including dysmorphic feature, congenital heart defect, immunodeficiency, hypoparathyroidism, learning and psychatric disorders [19]. Echo for FA confirmed that he has small ventricular septal defect (VSD) and unlikely the cause for his myocardial dysfunction. Moreover the repeated Echo on follow up showed stable VSD and most likely will close spontaneously.
This baby responded dramatically to treatment with alfacalcidol and calcium, and cardiac function returned to normal within months.
FA is 6 years old now on regular follow up in the endocrine and genetic clinic. His cardiac function remained normal, evaluated at one and half years after initial presentation. His calcium level is monitored every 4-6 months and remained above 2.00 mmol/L. He is on alfacalcidol maintenance dose. During follow up he was noticed to have motor and cognitive delay so referred to responsible teams.
Conclusion
The exact mechanisms of this hypocalcemia induced DCM and how reversal can be achieved with prompt treatment of hypocalcemia and underlying cause remain unclear. However, this association is of noted importance when encountering a patient with a new diagnosis of DCM. With increased suspicion, the diagnosis of hypocalcemia induced DCM can be made and prompt treatment can lead to improved cardiac function and better patient outcome. We believe that this is first case of congenital hypoparathyrodism-confirmed DiGeorge syndrome presenting with dilated cardiomyopathy induced by hypocalcaemia.
References
- Cox GF, Sleeper LA, Lowe AM, Towbin JA, Colan SD, Orav EJ, et al. Factors associated with establishing a causal diagnosis for children with cardiomyopathy. Pediatrics. 2006; 118: 1519-1531.
- Kim BG, Chang SK, Kim SM, Hwang JS, Jung JW. Dilated Cardiomyopathy in a 2 Month-Old Infant: A Severe Form of Hypocalcemia With Vitamin D Deficient Rickets. Korean Cric J. 2010; 40: 201-203.
- Gulati S, Bajpai A, Juneja R, Kabra M, Bagga A, Kalra V. Hypocalcemic heart failure masquerading as dilated cardiomyopathy. Indian J Pediatr. 2001; 68: 287-290.
- Avramides DA, Ionitsa SS, Panou FK, Ramos AN, Koutmos ST, Zacharoulis AA. Dilated Cardiomyopathy and Hypoparathyroidism: Complete Recovery after Hypocalcemia Correction. Hellenic J Cardiol. 2003; 44: 150-154.
- Abdullah M, Bigras JL, McCrindle BW. Dilated cardiomyopathy as a first sign of nutritional vitamin D deficiency rickets in infancy. Can J Cardiol. 1999; 15: 699-701.
- Sung JK, Kim JY, Ryu DW, Lee JW, Youn YJ, Yoo BS, et al. A case of hypocalcemia-induced dilated cardiomyopathy. J Cardiovasc Ultrasound. 2010; 18: 25-27.
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002; 415: 198- 205.
- Jayakumar A, Cheng L, Liang CT, Sacktor B. Sodium gradient-dependent calcium uptake in renal basolateral membrane vesicles. Effect of parathyroid hormone. J Biol Chem. 1984; 259: 10827-10833.
- Brown J, Nunez S, Russell M, Spurney C. Hypocalcemic Rickets and Dilated Cardiomyopathy: Case Reports and review of literature. Pediatr Cardiol. 2009; 30: 818-823.
- Abdullah M, Bigras JL, McCrindle BW. Dilated cardiomyopathy as a first sign of nutritional vitamin D deficiency rickets in infancy. Can J Cardiol. 1999; 15: 699-701.
- Price DI, Stanford LC Jr, Braden DS, Ebeid MR, Smith JC. Hypocalcemic Rickets: An Unusual Cause of Dilated Cardiomyopathy. Pediatr Cardiol. 2003; 24: 510-512.
- Tomar M, Radhakrishnan S, Shrivastava S. Myocardial dysfunction due to hypocalcemia. Indian Pediatr. 2010; 47: 781-783.
- Breslau NA, Pak CY. Hypoparathyroidism. Metabolism. 1979; 28: 1261-1276.
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002; 415: 198- 205.
- Giles TD, Iteld J, Rives KL. The cardiomyopathy of hypoparathyroidism. Another reversible form of heart muscle disease. Chest. 1981; 79: 225-225- 229.
- Giles TD, Iteld BJ, Rives KL. The cardiomyopathy of hypoparathyroidism. Another reversible form of heart muscle disease. Chest. 1981; 79: 225-229.
- Tonelli AR, Kosuri K, Wei S, Chick D. Seizures as the first manifestation of chromosome 22q11.2 deletion syndrome in a 40-year old man: a case report. J Med Case Reports. 2007; 1: 167.
- Restivo A, Sarkozy A, Digilio MC, Dallapiccola B, Marino B. 22q11 Deletion syndrome: a review of some developmental biology aspects of the cardiovascular system. J Cardiovasc Med. 2006; 7: 77-85.
- Debbané M, Glaser B, David MK, Feinstein C, Eliez S. Psychotic symptoms in children and adolescents with 22q11.2 deletion syndrome: Neuropsychological and behavioral implications. Schizophr Res. 2006; 84: 187-193.