
Case Report
J Pediatr & Child Health Care. 2022; 7(1): 1055.
Clinical and Biological Characteristics of a Griscelli Syndrome Type 2: About a Case Report
Nahi C*, Abdessadek S, Benouachane T and Bentahila A
Pediatric Cardiology, Rheumatology and Nephrology Department, Rabat Children’s Hospital/Morocco
*Corresponding author: Nahi C, Pediatric Cardiology, Rheumatology and Nephrology Department, Rabat Children’s Hospital/Morocco
Received: September 19, 2022; Accepted: October 20, 2022; Published: October 27, 2022
Abstract
Introduction: Griscelli Syndrome (GS) is a rare autosomal recessive genetic disorder, characterized by a partial albinism that can be associated with early-onset severe psychomotor retardation, and normal immune status (type 1), or associated with an immune deficiency (type 2). GS type 2 results from mutations in the RAB27A gene, leading to immune deficiency. GS type 2 has a grave prognosis because of a rapidly fatal macrophage activated syndrome.
Case Report: A 3-month-old infant having a family history of first-degree consanguinity in the parents and history of silvery gray hair in two uncles, Admitted to our department for management of a prolonged fever and an anemic syndrome. Hair on the scalp, eyebrows, and eyelashes were silvery gray and had a metallic sheen. The lymphocyte sub typing showed a severe combined immune deficiency. Histological examination of the hair showed enlarged, hyper pigmented basal melanocytes with sparse pigmentation of adjacent keratinocytes in favor of a Griscelli syndrome. The evolution was marked by clinical worsening, respiratory distress. On the biological level, a macrophage activation syndrome was retained in front of a hyperferritinemia, a hypertriglyceridemia and hepatic cytolysis. The patient received broad-spectrum antibiotic therapy, antivirals and antifungals, corticosteroid boluses 3 days in a row and an immunoglobulin infusion. Unfortunately, the patient died of septic shock.
Conclusion: Type 2 GS is a rare genetic disease with a poor prognosis, which must be diagnosed early to allow better support. The only curative treatment is bone marrow transplantation.
Keywords: Griscelli syndrome; genetic; Immune deficiency; Macrophage activation syndrome
Introduction
Griscelli Syndrome (GS) is a rare autosomal recessive genetic disorder, characterized by apartial albinism resulting from dilution pigmentation of hair and skin [1]. Three genes are currently identified in the control of intramelanocyte transport of melanosomes and in their transfer to keratinocytes. An association with neurological disorders or an immune deficiency respectively defines the SGs of type 1 and type 2. In contrast, SG type 3 is a form isolated with exclusively cutaneous expression. We report a case of Griscelli syndrome type 2 diagnosed at the age of 3 months revealed by macrophage activation syndrome.
Case Report
A 3-month-old infant, second of two siblings. Admitted to our department for management of a prolonged fever and an anemic syndrome. The child had a family history of first-degree consanguinity in the parents and history of silvery gray hair in two uncles. Moreover, his brother is doing well. At birth, he was noticed to have silvery gray hair with a metallic sheen. There was no delay in the achievement of this child’s developmental milestones. The history of the disease goes back to twenty days before his admission by the installation of an unquantified fever and vomiting, all aggravated by mucocutaneouspallor, which motivated a biological assessment, requested externally objectifying a pancytopenia.
The clinical examination on admission finds an active child, weighing 6 kg, pale, hemodynamically and respiratory stable. Hair on the scalp, eyebrows, and eyelashes were silvery gray and had a metallic sheen (Figure 1). The color of the skin was light brown and lighter than the mother’s skin color, whereas the iris was normal. Abdominal, pleuro-pulmonary Neurologic, cardiovascular, and ophthalmologic examinations were initially normal.
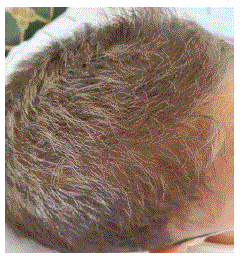
Figure 1: Silvery gray Hair on the scalp having a metallic sheen.
Biologically, the blood count shows pancytopaenia with normochromic normocytic are generative anemia at 4.7 g/dl, Leukoneutropenia (White Blood Cells at 2920/mm3 and neutropenia at 230/mm3), Thrombocytopenia at 8000/mm3. The smear shows erythrocyticanisopoikilocytosis. Medulogram came back normal. The inflammatory assessment shows a CRP mg/L at 122 with a hyperferritinemia at 6090 μg/l. Blood cultures came back negative. The lymphocyte sub typing showed a severe combined immune deficiency (CD3 T lymphocytes at 193 / μl, CD4 T at 91/μl, CD8 T at 87/μl, B-lymphocytes CD19+ at 4/μl, NK cells at 29/μl). A standard lung X-ray was requested which returned without any particularities.
Histological examination of the hair showed enlarged, hyperpigmented basal melanocytes with sparse pigmentation of adjacent keratinocytes in favor of a Griscellisyndrome (Figure 2). Genetic testing could not be done.
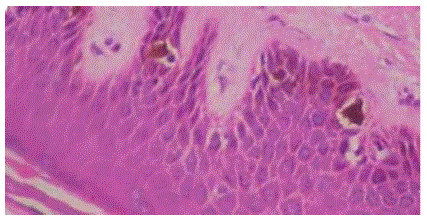
Figure 2: Histological examination of the hair showed enlarged, hyper
pigmented basal melanocytes with sparse pigmentation of adjacent
keratinocytes in favor of a Griscelli syndrome (Hematoxylineosin stain;
original magnification X4.).
He was treated with intravenous cefotaxime, amikacin, and vancomycin. He also received an immunoglobulin infusion.
The evolution was marked by clinical worsening, respiratory distress. The clinical examination shows a pleural effusion syndrome with dullness on palpation, edema of the lower limbs and ecchymotic purpura affecting the whole body. On the paraclinical level, the chest X-ray objectified a left pleural effusion and an atelectasis of the left lung (Figure 3). On the biological level, a macrophage activation syndrome was retained in front of a hyperferritinemia at >40000 μg/l, a hypertriglyceridemia at 3.5 4 g/L, hepatic cytolysis (ALAT at 328). Abdominal ultrasound showed homogeneous hepato-splenomegaly with peritoneal effusion blade. Chest CT scan showed marked left bilateral pleural effusion with left atelectasis.
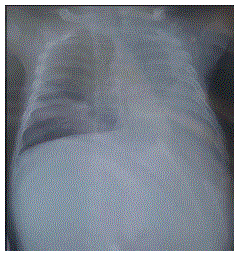
Figure 3: Chest X-ray showing a left pleural effusion and an atelectasis of
the left lung.
The patient received broad-spectrum antibiotic therapy, antivirals and antifungals, corticosteroid boluses 3 days in a row and an immunoglobulin infusion. Unfortunately, the patient died of septic shock.
Discussion
The GS is a rare genetic disease, of autosomal recessive transmission, which affects both men and women. It was first described in 1978 by Griscelli et al. who reported a syndrome associating partial albinism with immunodeficiency in two patients with silvery gray hair and frequent pyogenic infections [3]. It is characterized by partial albinism and a silver sheen of the hair, and it is now classified into 3 types on the basis of genetic and molecular features. GS type 3 represents patients with isolated silvery gray hair, light-colored skin. This can be associated with early-onset severe psychomotor retardation, and normal immune status (type 1), or associated with an immune deficiency (type 2) [3]. Since its first description, more than 60 cases have been reported, the majority of which come from the Mediterranean region, especially in Turkey [4] in addition to a few rare cases in India [5].
Each type of SG is secondary to mutations in a different gene. GS type 1 is caused by mutations in the myosin Va gene (MYO5A) located on chromosome 15q21, which regulate organelle transport in both melanocytes and neuronal cells explaining the neurological symptomatology [4]. GS type 2 results from mutations in the RAB27A gene located on chromosome 15q21, less than 1.6 cM from the MYO5A gene [6]. The RAB27A protein a guanosine triphosphatase that plays an essential role in peripheral transport of melanosomes to the neighboring cells such as keratinocytes and granule exocytosis in cytotoxic T lymphocytes [6]. Abnormalities of RAB27A are responsible for neutropenia and NK cell dysfunction leading to immune deficiency and an abnormal cytotoxic response that can lead to a syndrome of uncontrolled activation of T cells and macrophages, known as hemophagocytic syndrome [6]. Hemophagocytic syndrome is characterized by infiltration of lymph nodes and other organs (including the brain) by polyclonal, activated T cells, mostly of the CD8 subset, and by activated macrophages that phagocytize blood cells.
SG is characterized by the presence of large aggregates of pigment in the hair sheath, visible under an optical microscope [3]. Cutaneous melanocytes contain many irregularly shaped melanosomes with incomplete blockade of transfer to surrounding keratinocytes [3]. The main differential diagnosis is the Chediak-Higashi syndrome, which is characterized clinically by the association of partial albinism and an immune deficiency, and biologically by the presence of giant intracytoplasmic granules in polymorphonuclear cells and azurophilic granulations in lymphocytes [7]. Other syndromes associating albinism and immunodeficiency can be confused with GS such as Hermansky-Pudlak syndrome type 2 (AP3B1 gene mutations) and p14 deficiency (MAPBPIP gene mutations) [7].
In our case, the diagnosis of GS type 2 was made before a set of clinical and biological arguments:the consanguineous character of the family, the history of repeated infections, the absence of psychomotor retardation, the oculo-cutaneous hypopigmentation, the silver reflection of the hair, the occurrence of SAM following a community lung infection and above all the Pathognomonic appearance of hair on microscopic examination. The absence giant granules in the nucleated cells ruled out Chediak-Higashi syndrome.
GS type 2 is characterized by an immune deficiency responsible for repeated infections. It constantly evolves towards a so-called “acceleration” phase, which manifests as a rapidly fatal macrophage activated syndrome [8]. Patients with GS type 2 may present with neurological symptoms secondary to infiltration of the central nervous system by activated hematopoietic cells.
Concerning the prognosis the means of treatment, in patients with GS type 1, there is no definitive cure, and the outcome is guarded as it depends on the severity of neurologic manifestations. Patients should receive supportive care for primary neurologic symptoms such as hypotonia and developmental delay, and seizures should be controlled adequately when present. GS type 2 has a grave prognosis, chemotherapy (VP16) or more recently anti lymphocyte serum and ciclosporin can be used to delay the hyper activation of lymphocytes and macrophages, and reduce symptoms attributable to organ infiltration. Intrathecal injections of methotrexate are effective in cerebral invasions. Currently, allograft bone marrow remains the only treatment allowing complete and prolonged remission [8,9]. Genetic counseling should be offered to families with affected children. Prenatal diagnosis of GS can be performed by light microscopic examination of hair from fetal scalp biopsy specimens and also by molecular genetic studies of fetal DNA, if the mutation is known [10].
Conclusion
Type 2 GS is a rare genetic disease with a poor prognosis, which must be diagnosed early to allow better support. The only curative treatment is bone marrow transplantation.
References
- Griscelli C, Durandy A, Guy-Grand D, Daguillard F, Herzog C, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med.1978; 65: 691-702.
- Dessinioti C, Stratigos AJ, Rigopoulos D, Katsambas AD. A review of genetic disorders of hypopigmentation: lessons learned from the biology of melanocytes. Exp Dermatol. 2009; 18: 741-749.
- Klein C, Philippe N, Le Deist F, Fraitag S, Prost C, et al. Partial albinism with immunodeficiency (Griscelli syndrome). J Pediatr. 1994; 125: 886-995.
- Manglani M, Adhvaryu K, Seth B. Griscelli syndrome - a case report. Indian Pediatr. 2004; 41: 734-737.
- Cagdas¸ D, Ozgür TT, Asal GT, Tezcan I, Metin A, et al. Griscelli syndrome types 1 and 3: analysis of four new cases and long-term evaluation of previously diagnosed patients. Eur J Pediatr. 2012; 171: 1527-1531.
- Meeths M, Bryceson YT, Rudd E, Zheng C, Wood SM, et al. Clinical presentation of Griscelli syndrome type 2 and spectrum of RAB27A mutations. Pediatr Blood Cancer. 2010 ; 54 : 563-572.
- Rezaei N, Moazzami K, Aghamohammadi A, Klein C. Neutropenia and primary immunodeficiency diseases. Int Rev Immunol. 2009; 28: 335-366.
- Pachlopnik-Schmid J, Moshous D, Boddaert N, Neven B, Dal Cortivo L, et al. Hematopoietic stem cell transplantation in Griscelli syndrome type2:a singlecenter report on 10 patients. Blood. 2009; 114: 211-218.
- Aricò M, Zecca M, Santoro N, Caselli D, Maccario R, et al. Successful treatment of Griscelli syndrome with unrelated donor allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2002; 29: 995-998.
- Durandy A, Breton-Gorius J, Guy-Grand D, Dumez C, Griscelli C. Prenatal diagnosis of syndromes associating albinism and immune deficiencies (Chediak-Higashi syndrome and variant). Prenat Diagn. 1993; 13: 13-20.