
Abstract
Approximately 75% of the drugs that are being approved for use in the pharmaceutical industry have low aqueous solubility. This leads to issues with drug uptake, higher doses and consequently a negative environmental impact. One approach to improving these Active Pharmaceutical Ingredients (APIs) is to use the crystal engineering technique of adding another molecule into the crystal lattice of the API. The formation of a novel solid-state is dependent on the strength of the intermolecular bonds formed and the type of synthesis used. Main synthesis routes include a variety of solution crystallization procedures as well as Liquid Assisted Grinding (LAG) and neat grinding. This review will emphasize the formation of new states by adding a distinct molecule into the lattice of the API through non-ionic bonding. For this reason, polymorphs and salts will be disregarded and the main forms analyzed are co-crystals, solvates, eutectics, and co-amorphous materials. Appropriate choice of excipient as well as screening methods is also given in detail.
Keywords: Review; Co-amorphous; Co-crystals; Salts/salt selection; Solid solutions; Solid-state; Solvates
Introduction
Every year the Food and Drug Administration (FDA) approves New Molecular Entities (NMEs) for pharmaceutical use. As of May 2017, there have already been 20 Active Pharmaceutical Ingredients (APIs) accepted, this year. A Biopharmaceutics Classification System (BCS), depicted in Figure 1, is used by the FDA to classify APIs and reduce unnecessary human testing [1]. The BSC classification is as follows: Class I (high permeability, high solubility); Class II (high permeability, low solubility); Class III (low permeability, high solubility); and Class IV (low permeability, low solubility) [2]. When combined with dissolution studies, a biowaiver for in vivo bioavailability and bioequivalence can be applied for Class I and III drugs, which leads to a less vigorous testing required for these drugs [3].
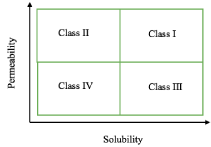
Figure 1: BCS used by the FDA where highly soluble is considered when the
highest dose strength is soluble in less than 250mL water over a pH range of
1 to 7.5, and highly permeable is considered when the extent of absorption
in humans is determined to be greater than 90% of an administered dose [1].
A simple solution to help pharmaceutical companies with more efficient drug testing (i.e. less clinical testing) would be to stop using drugs that do not belong to Class I and III; however, approximately 75% of NMEs belong to BCS Class II and IV [4]. This means that the majority of effective medicines that are FDA approved drug innovations have a poor solubility in aqueous solutions. Many drugs today are designed using high throughput screening approaches to ensure valuable and successful products. Since the approval process for NMEs takes years, pharmaceutical companies want to guarantee that they have the best contenders. The screening processes tend to result in APIs that are higher in molecular weight and have increased hydrogen-bonding [5]. Increased molecular size tends to result in greater van der Waals interaction between like molecules, while more hydrogen bonding within the API creates a stronger crystal lattice. Both characteristics lead to a compound that is less soluble.
Why are low solubility drugs undesirable? First, they often require high doses to reach required therapeutic plasma concentrations after oral administration, hence patients are ingesting significantly more than necessary. The increase in dose also leads to more costly medicines, which is unfavorable to consumers and pharmaceutical companies. Poorly soluble APIs are also more vulnerable to food effects (e.g. some drugs are suggested to be taken after a fasting period due to reduced absorption in the presence of food), and slow dissolution can lead to lower fraction absorbed in the small intestine [6]. High levels of API excretion due to low absorption are costly to the pharmaceutical industry and can be detrimental to our environment. For instance, Carbamezapine, a routine API that is nearly insoluble in water, was found in every sample taken from wastewater treatment plants’ effluent of three different rivers in England [7]. Therefore, not only are Class II and IV drugs more expensive to administer, they also have a lasting effect on the environment. Taking these factors into account, a strong need to increase the solubility of these drugs is prevalent.
Crystal engineering is defined as: the understanding of intermolecular interactions in the context of crystal packing and the utilization of such understanding in the design of new solid forms with desired physical and chemical properties [8]. Numerous methods are present that involve altering the solid-state of Class II and IV APIs to increase water solubility and bioavailability. This altering of states is known as crystal engineering, and some of these approaches include formation of: salts, co-crystals, solvates, eutectics and co-amorphous mixtures. The bioavailability of a drug is affected by its aqueous solubility, dissolution rate, permeability, etc. If one can increase the water solubility of Class II drugs, the bioavailability will be greatly improved [9].
In this review, the practices discussed will focus on non-ionic bonding between distinct molecules, so salts and polymorphs will be overlooked. The techniques mentioned involve adding another component called an excipient into the crystal structure; excipients will usually have more ideal physical and chemical characteristics than the API, so the combination will lead to a superior drug formulation. The beauty of these methods relies on the fact that excipients are already approved by the FDA for use in pharmaceuticals, and thus, the novel solid-state of the API will not need to undergo new clinical studies.
Co-amorphous Mixtures
An amorphous solid, also referred to as a glass, has no long range molecular order of packing nor do its constituents have a distinct molecular conformation [10]. Amorphous materials have properties similar to liquids at a molecular level but are more like solids at a macroscopic level [11]. Due to the lack of order and since this form is the most energetic, amorphous compounds have been shown to have increased solubility in comparison to their crystalline counterparts [12]. Consequently, this will also result in an increase in bioavailability, so the formation of an amorphous pharmaceutical compound is an appropriate means to tackle the issues with Class II and IV drugs.
Co-amorphous solids, depicted in Figure 2, occur when two components become amorphous due to interaction with one another. Intermolecular bonding between API and excipient impedes the arrangement of individual molecules into separate lattices and results in an amorphous material [13]. Co-amorphous mixtures are shown to be not only high in solubility, but also have a high stability [14]. By adding another component into the system, the problem of low stability in amorphous materials is alleviated in co-amorphous materials. Amino acids are great candidates as excipients in these systems [15].
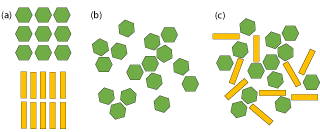
Figure 2: Representation of (a) crystalline API (hexagon) and excipient
(rectangle), (b) amorphous API, and (c) co-amorphous API and excipient.
An amorphous phase composed of a single material can be created through two main routes of production: either a thermodynamic or kinetic disordering process. Thermodynamic disordering occurs when the loss of long range order symmetry causes disorder in the solid-state. It allows for close random packing and is termed thermodynamic disorder due to the breaking of symmetry [16]. Kinetic disordering is referred to when long range disorder is caused by short range order. This process is considered to be continuous and can reduce crystalline material to glassy material [17]. When creating an amorphous state, the thermodynamic pathway involves starting with a thermodynamically stable non-crystalline form, such as a melt or solution. The melt will then undergo quenching, while the drug will precipitate from solution. Conversely, the kinetic pathway will start with a solid-state which is then converted by introducing crystal defects via crushing, milling or continuous shear forces [18]. These pathways are also applicable to the preparation of co-amorphous systems.
While still quite a new practice in drug formulations, coamorphous mixtures have been shown to be readily obtained by a variety of procedures. The main procedures are milling and grinding (kinetic disordering) or quenching/cooling and solvent evaporation (thermodynamic disordering). Given in Table 1 are some instances of co-amorphous mixtures and the methods used to produce them. These are all desirable techniques because not only are they quick to produce, they also only require a small amount of sample; however, not every technique is applicable to every drug-excipient combination. Quenching is not available if degradation occurs at higher temperatures, solvent evaporation can prove difficult if there are solubility differences, and milling may not apply enough force to disrupt the lattice.
![]()
Drug
Excipient
(or drug)
Technique
Experimental
Curcumin
Artemisinin
Solvent Evaporation
A mixture of 1:1 molar ratio of Curcumin and Artemisinin was dissolved in ethanol. The solution was then rotovaped (fast evaporation under vacuum) to give the co-amorphous product [19].
β-azelnidipine
Maleic Acid
Grinding (LAG or neat)
β-azelnidipine and maleic acid at molar ratios of 1:1 and 1:2 were placed into a 50mL stainless steel jar and milled at 30Hz in a laboratory-scale oscillatory disk mill. This can be done neat or with the addition of ethanol [20].
Indomethacin
Cimetidine
Quenching
Indomethacin–cimetidine physical mixtures were melted on an aluminium foil placed on a preheated hotplate (180°C) for 1 min. The molten liquid was cooled quickly by placing the aluminum foil over ice water [21].
Table 1: Preparation of Co-amorphous Systems.
To classify a material as amorphous, perhaps the most effective method is to use powder X-ray diffraction (PXRD). Crystalline material will show very distinct and sharp peaks, while amorphous material will appear as one very broad peak termed a halo since it has no defined crystal lattice or symmetry operators. To further examine the characterization of a co-amorphous system, the first example in Table 1 will be considered. Curcumin, a poorly water-soluble molecule, has been shown to help increase anti-malaria effects when taken in parallel with the API Artemisinin. Instead of prescribing these and taking them separately, a co-amorphous mixture was formed via fast solvent evaporation so that a patient could take them together. The PXRD patterns of the starting materials and the mixture are shown in Figure 3 [19]. It is clear that the mixture has lost the crystalline nature of the starting materials and is now forming weak intermolecular interactions.
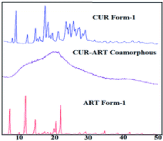
Figure 3: The PXRD patterns of crystalline Curcumin form I (CUR Form-I),
co-amorphous Curcumin-Artemisinin (CUR-ART Co-amorphous), and
crystalline artemisinin form I (ART Form-I) [19].
Depending on the method used for producing the co-amorphous mixture, obtaining an amorphous pattern from PXRD is sufficient for characterization. If prepared via grinding, then the product is usually co-amorphous; however, quenching may lead to degradation and solvent evaporation could lead to a single amorphous state precipitating out first. To ensure that both starting materials are present in the amorphous product, one should preform solution HPLC or 1H NMR (nuclear magnetic resonance). In the NMR analysis, if the 1H shifts seen are solely representative of both starting materials, then the product can be said to be co-amorphous. Keeping with the Curcumin-Artemisinin mixture, it was exhibited that chemical shifts for both starting materials were present in their product.
Solid-state NMR (ssNMR) is another method that can be used for the characterization of these systems. ssNMR will usually show broader bands for amorphous material compared to crystalline material due to the increase of molecular orientations [22]. There will also be a shift in peaks due to the intermolecular bonding between the drug and excipient [23]. Furthermore, when compared to the ssNMR spectrum of a purely crystalline homogeneous mixture of the starting materials, the amorphous content of the co-amorphous system can be determined [24]. However, this technique often relies on peak integration, which can prove to be difficult when there is an overlap (i.e. broad peaks in amorphous materials often result in an overlapping signals). Also, formation of the homogeneous mixture could result in decreased crystallinity that would introduce error. Another method is to compare with both the fully crystalline and fully amorphous spectra of the starting materials. This method uses Principal Components Analysis (PCA) of ssNMR data that is normalized by dividing all PCA scores by the score of the 100% amorphous sample after subtracting the score of the crystalline sample. The profile for rate of co-amorphization can be obtained by plotting the normalized data against mill time [25].
For further characterization, Raman and FT-IR (Fourier Transform Infrared Spectroscopy) can be conducted. For both techniques, the spectra will appear as a combination of the starting materials with small shifts in frequencies [26]. The peaks that have shifted when compared to the starting materials, are representative of the bonds involved in the intermolecular bonding that stabilizes and causes formation of co-amorphous mixtures [27,28]. When coupled with ssNMR, these characterization techniques are useful in proposing a bonding mechanism between components of the system. Following the ongoing example, the proposed bonding mechanism of Curcumin and Artemisinin in Figure 4 was determined by analyzing the FT-IR spectrum, which showed a shift in carbonyl stretching of Artemisinin and the phenolic alcohol of Curcumin.
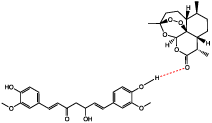
Figure 4: Proposed bonding mechanism in the co-amorphous system of
Curcumin-Artemisinin [19].
Fundamental thermal analysis of these systems can be carried out with Differential Scanning Calorimetry (DSC). Amorphous materials will undergo a glass transition at a specified temperature (Tg), above which the material will tend to crystallize [29]. The DSC data of a co-amorphous system will differ vastly from that of the starting materials. Once again, considering the Curcumin-Artemisinin system, the differences can be appreciated in Figure 5. The melting of the starting materials is seen in the endothermic peaks, while the co-amorphous system has a broad exotherm. The Tg is highlighted and appears below the melting of both Curcumin and Artemisinin. When plotting a phase diagram of a binary system using DSC, a “v” shape may appear for co-amorphous mixtures with a single thermal event happening at a particular molar ratio [30]. Since co-amorphous systems are not well studied, using DSC alone is not useful as its results are not universal (e.g. the formation of a “v” in the binary phase diagram is an ideal situation).
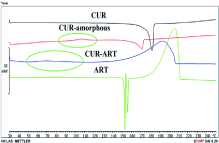
Figure 5: DSC thermographs of Curcumin (CUR), amorphous Curcumin
(CUR-amorphous), co-amorphous Curcumin-Artemisinin system (CUR-ART)
and Artemisinin (ART). Circled are the glass transition temperatures.
Co-Crystals
Much like co-amorphous systems, a co-crystal is composed of neutral, solid components that interact with each other through intermolecular bonding. A simple depiction of a 1:1 molar ratio cocrystal comprised of two different compounds is given in Figure 6. In this representation, the hexagonal component can represent the API, while the rhombus is the excipient. Intriguingly, individual components can be salts or hydrates so long as each is considered overall neutral [31,32]. Co-crystals have been known for some time by different names (e.g. addition compounds, mixed binary molecular crystals, molecular complexes, solid-state complexes, heteromolecular crystals, etc.), but have not been of great interest until recently [33]. To avoid being ambiguous between solid-states, the FDA gave the following definition to co-crystals: solids that are crystalline materials composed of two or more molecules in the same crystal lattice [34]. In a pharmaceutical co-crystal, at least one of these components will be an API; the excipient is referred to as the cocrystal former or co-former [35].

Figure 6: Representation of a two-component co-crystal in a 1:1 molar ratio,
where both hexagons and rhombuses represent a solid, neutral component.
Co-crystallization occurs when an excipient is introduced that competes with and supersedes the intermolecular bonding of a molecule with itself [36]. The most difficult task in co-crystal formation is picking an appropriate co-former, which will be discussed later on. The main intermolecular force behind co-crystal formation is hydrogen bonding, so the co-former should have appropriate Hydrogen Bond Donors (HBD) and Acceptors (HBA). There are additional contributing factors to co-crystal formation, and a statistical review of co-crystals in the CSD (Crystal Structure Database) leads to some general assumptions: co-crystals form between molecules of similar shape and polarity, and the number of HBD/HBA is less important than the strength of the potential hydrogen bonding [37]. For instance, if an API has multiple functional groups and one is carboxylic, it will tend to have greater potential for forming a co-crystal with an excipient containing an amide than with an excipient that can form multiple weaker bonds. Furthermore, if an API shows potential for intermolecular bonding with an excipient due to matching the HBD and HBA groups, a co-crystal may not form if the shapes do no match (e.g. planar molecules will tend to form co-crystals with planar molecules).
The formation pathways for co-crystallization are comparable to those for co-amorphous mixtures and include Liquid Assisted Grinding (LAG) and dry/neat grinding, solvent evaporation, isothermal slurry conversion, cooling crystallization, anti-solvent crystallization, spray drying and rapid expansion of supercritical fluids. Provided in Table 2 is a summary which highlights examples of each method.
![]()
Drug
Co-former
Technique
Experimental
Theophylline
Citric Acid
Neat Grinding
0.150g of an equimolar combination of theophyllinecitric acid and citric acid was placed into a 25mL stainless steel grinding jar and ground for 1h using two stainless steel balls 7mm in diameter. The overall temperature of the reaction mixture remained below 35°C [38].
Adefovir Dipivoxil
Glutaric Acid
LAG
1:1 mole ratio of Adefovir Dipivoxil and Glutaric Acid was ground using an agate mortar and pestle for 20-30 min. Methanol was added dropwise over the course of grinding (0.5mL/mmol cocrystal) [39].
Fenofibrate
Nicotinamide
Solvent Evaporation
A mixture of FNO and nicotinamide in 1:1 molar ratio was dissolved in 20mL of ethanol with sonication (to induce saturation) and was kept overnight to evaporate solvent. The crystals obtained after evaporation of ethanol were dried in air [40].
Trospium Chloride
Salicylic Acid
Quenching
A 1:1 physical mixture of Trospium Chloride with salicylic acid was prepared and heated at 10°C per min under nitrogen purge above eutectic temperature. The mixture was then cooled to room temperature at a cooling rate of 10°C per min. After the mixture stood at 30°C for 60 min, the heating was repeated and the co-crystal was obtained [41].
Indomethacin
Saccharin
Anti-solvent Addition
In a 1L double jacketed reactor, predetermined amounts of Indomethacin and saccharin were fully dissolved in methanol (300mL), and 150mL of purified water as anti-solvent was added to the solution using a peristaltic pump. The process was performed for 30 min and the product was filtered and dried under vacuum.
Theophylline
Glutaric Acid
Slurry Conversion
Equimolar mixture of theophylline and glutaric acid was added into chloroform to form slurry at 50°C. This slurry was seeded with crystals from co-grinding of theophylline and glutaric acid and co-crystals formed as solvent evaporated.
Theophylline
Saccharin
Spray Drying
A 0.9% equimolar solution of Theophylline and saccharin in ethanol were spray dried at an inlet temperature of 120°C and outlet temperature of 77°C to give a co-crystal. The feed rate was 5mL/min, the air flow was 536L/h and the aspiration was 100% [42].
Table 2: Some Preparation Techniques for Co-Crystal Synthesis.
The most advantageous way to characterize a co-crystal is by growing single crystals and running X-ray diffraction. This technique will give the stoichiometric ratio, crystal lattice and intermolecular bonding of the molecular structure [44-47]. Single crystal X-ray diffraction alone is adequate for identifying co-crystal formation. However, while it is the most useful, obtaining single crystals is often challenging and not always possible. Since many co-crystals are formed using mechanochemical techniques, access to single crystals is often not possible [48]. Also, changing to conditions necessary to form high quality crystals can sometimes cause components to crystallize independently. In these scenarios, other techniques can be employed to characterize the suspected co-crystal.
ssNMR is a practical characterization technique in helping elucidate the structure of co-crystals. It has even been shown that ssNMR can be used to monitor co-crystal formation in situ [49]. This method can be used for small amounts of sample, is noninvasive and can adequately analyze multiphase systems [50]. Using 1D and 2D ssNMR techniques can lead to the determination of the intermolecular bonding sites within co-crystals [51]. When coupled with PXRD and Density Functional Theory (DTF) calculations, 1D ssNMR can be employed to determine the molecular structure of a co-crystal. For example, a theophylline-nicotinamide co-crystal has been formed by LAG or slow evaporation with ethanol and is shown to have improved solubility and hygroscopicity compared with the anhydrous API [52]. Characterization techniques and crystal engineering knowledge can be used to interpret intermolecular bonding sites in the co-crystal. Next, this suggested bonding between the API and excipient was refined using DTF method, and the 13C and 15N ssNMR spectra were predicted. These were then compared to the spectra that were obtained experimentally and validated the refined structure of the theophylline-nicotinamide co-crystal [53].
PXRD is also particularly useful; if a co-crystal is formed then the crystal lattice will change, resulting in a powder pattern that is distinct from the starting materials. This concept can be easily visualized by examining the theoretical PXRD patterns of an API, coformer and their corresponding co-crystal in Figure 7. It has also been demonstrated that PXRD can be utilized to help determine the purity of the co-crystal formed. This is done by formation of a calibration curve using known amounts of co-crystal and then monitoring the most intense peak in the PXRD pattern that has the least amount of overlapping [54].
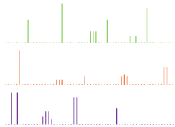
Figure 7: Theoretical PXRD patterns of (a) API (b) co-former and (c) their
co-crystal.
Thermal analysis is also critical in the analysis of co-crystals [55]. Akin to the PXRD patterns, DSC thermographs will result in data that is different from the starting materials. The thermographs of co-crystals usually show an endothermic peak for co-crystal melting that is above or below the melting of the API and co-former [56]. Ideally, the co-crystal will have a lower melting point than the API; this will result in intramolecular bonds that are easier to break and increase the drug’s aqueous solubility [57]. DSC can also be employed to prepare a phase diagram for the system. When plotting the onset of peak one and the peak temperature of peak two at varying molar ratios, the result is a sort of “w” shape [58]. The region between the two eutectic peaks is the co-crystal forming region. This phenomenon is more suitably explained with the aid of an image (Figure 8) [59].
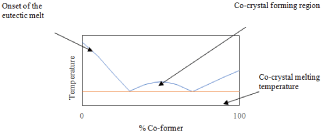
Figure 8: Theoretical phase diagram of a binary system capable of co-crystal
formation.
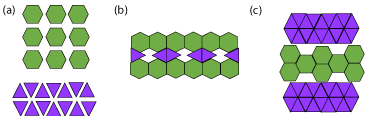
Figure 9: (a) molecular structures of API (hexagon) and excipient (triangle),
(b) and (c) are both examples of discontinuous interaction between API and
excipient found in eutectic mixtures. These arrangements can interact with
each other throughout the mixture [67].
Obtaining PXRD and DSC data that validates the presence of a co-crystal is sufficient in claiming co-crystal formation. To acquire the stoichiometric ratio, integration techniques of solution 1H NMR can be employed. For example, if a single proton in the API is normalized to one and a chemical shift representing three protons in the excipient integrates to three, then the co-crystal ratio is 1:2 API: co-former. If single crystal XRD and/or ssNMR have been attained, then Raman and FT-IR are not necessary. However, if not attained, these techniques will show a change in stretching frequencies which will imply that certain bonds are involved in intermolecular bonding and help to determine what supramolecular synthons are being formed [60,61]. DSC can be coupled with FT-IR as a very quick and easy means of co-crystal confirmation. By comparing the changes in IR frequencies as a binary system is heated, one can confidently see where co-crystal formation occurs [62]. Often times a combination of all or multiple techniques that have been mentioned are used to ensure a complete characterization so that co-crystal formation is irrefutable.
Eutectics
Like co-crystals and co-amorphous mixtures, eutectics comprise of multiple components that are neutral at ambient conditions. A pharmaceutical eutectic mixture comprises of API(s) and excipient(s) that do not interact to form a novel compound, but will impede the crystallization of each other at certain molar ratios [63]. These interactions are short range [64]. Formation of eutectic mixtures is a useful method for weakening the intermolecular bonding of a crystalline material. This will reduce the melting point of a system, which will ultimately increase the solubility. The solubility is also increased due to the reduction in particle size (and consequently the increase in surface area) found in eutectics compared to the separate crystalline components [65], Figure 9 is a visualization to help comprehend the concept of eutectics: if the hexagon and rhombus characters shown in (a) are representative of the crystal structures of API and excipient, then (b) and (c) are ways in which they can interact. These different types of interactions will be found discontinuously within the eutectic mixture and can interact with each other [66]. The components of a eutectic mixture are miscible in the liquid state, but only slightly in the solid-state. At a eutectic composition, the API and excipient will crystallize out at the same time; while at any other molecular ratio one component will start to crystallize distinctly [67].
The synthesis of eutectic mixtures is comparable to that of other solid-states. Usually the same techniques are employed and will simply result in a eutectic instead of a co-crystal or co-amorphous mixtures due to the strength of the interactions between API and excipient. The most common techniques include melting, grinding and solvent evaporation; some examples are displayed in Table 3.
![]()
Drug
Excipient (or drug)
Technique
Experimental
Curcumin
Nicotinamide
Grinding
Curcumin and nicotinamide were taken in a stoichiometric ratio and subjected to solid-state grinding in a mortar-pestle for 15 min [68].
Acetaminophen
Caffeine
Melting
A stoichiometric mixture of Acetaminophen and caffeine was heated to a temperature 2°C above complete fusion of both compounds. The mixture was stirred, cooled to room temperature and gently ground (using a mortar and pestle) [69].
Aspirin
Paracetamol
Solvent Evaporation
A different proportion of Aspirin and Paracetamol was dissolved in a methanol:dichloromethane (3:1) solution; the solvent was evaporated rapidly under vacuum [70].
Table 3: Some Synthesis Techniques of Eutectic Mixtures.
Eutectic mixtures are an emblematic product when screening for pharmaceutical co-crystals; consequently, the same techniques are employed for characterization. The initial step is typically to run PXRD due to its capacity for rapid analysis. The PXRD pattern for a eutectic mixture differs from that of co-crystals and co-amorphous mixtures since they are characterized by a pattern that is a combination of the starting materials (similar to a physical mixture) as opposed to one that is unique [71]. This distinguishing characteristic is due to API and excipient retaining their molecular structures while still capable of intermolecular bonding.
Obtaining a PXRD pattern of this nature is not sufficient for eutectic confirmation as it may simply be a physical mixture of the components; the next step in characterization would be DSC. The thermograph for a eutectic mixture will usually appear as a melting endotherm that occurs at a temperature below both API and excipient melting [72]. A physical mixture differs and will show two distinct melting points, since the constituents are not interacting [73]. At a specific ratio, this endotherm will appear as a single peak resembling a co-crystal; however, since the PXRD pattern is not distinct, it is known to be a eutectic and no exothermic events will occur [58]. Running DSC at different molar ratios and plotting the binary phase diagram (as previously described) will result in a graph that has a “v” shape similar to Figure 10.
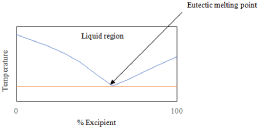
Figure 10: Theoretical phase diagram of a binary system which forms a
eutectic.
These two techniques, PXRD and DSC, are typically sufficient in determining if a eutectic has formed. Solution 1H NMR can be run to confirm stoichiometric ratios. FT-IR, Raman and ssNMR will all mirror PXRD in terms of obtaining result that reflect a combination of the starting materials.70 Confocal Raman Microscopy has been employed to show discontinuous bonding in eutectic mixtures by monitoring characteristic bonding peaks [74].
Solvates
Contrary to the previous solid-states that have been discussed and consist exclusively of solid materials at room temperature, a solvate comprises of multiple components where solvent(s) has been incorporated into the crystal lattice [75]. A special case, termed a hydrate, occurs when solvent involved is water [76]. Liquid solvent at ambient conditions can be found in both stoichiometric and nonstoichiometric amounts, as depicted in Figure 11 [77]. A solvate is described simply as any crystal composed of a solvent molecule and either a co-former or a cation-anion pair (Figure 11(c)). When the components are found in stoichiometric amounts, this is referred to as a “true solvate” (Figure 11(b)) [78].
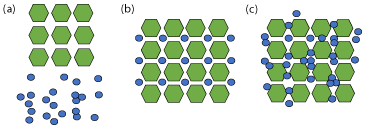
Figure 11: Representation of (a) crystalline API (hexagons) and solvent
(small circles), (b) stoichiometric or ‘true” solvate, and (c) non-stoichiometric
solvate.
Pharmaceutical solvates will contain an API as a co-former or pharmaceutical salt as the cation-anion pair. Solvent can incorporate into the voids of the crystal lattice and is recognized as channel solvent [79]. These types of solvates will retain their crystal structure if the solvent is removed and can be found in stoichiometric or non-stoichiometric amounts [80]. Another type involves stronger intermolecular bonding between the solvent and the solid that results in a distinct and more stable crystal lattice. In this scenario, functionality is a more vital factor in solvent inclusion [81]. The extent of stabilizing, intermolecular bonding found in these solvates includes: hydrogen bonding, halogen binding, metallic bonding and van Der Waals Interactions [82,83].
Since solvent (especially water) is present in many stages of pharmaceutical production, solvates are an interesting means of increasing the solubility and stability of drugs. Water and many other solvents have potential to form strong hydrogen bonding interactions with a variety of molecules that could result in the formation of a crystal lattice with increased stability. Removing solvent can lead to the decomposition of the API or ruin its crystal lattice [84]. Instead of trying to remove solvent from the system as waste, alternatively, it can be incorporated (so long as it is safe for ingestion).
Synthesis of solvates is usually achieved via cooling and/or evaporative crystallization from solution. Both methods rely on supersaturation to promote crystal growth. Solvates have also been formed via grinding [85]. A case for the synthesis of each type of solvate is given in Table 4.
![]()
Solvent
Solvate Type
Experimental
Isopropanol
Stoichiometric Channel
Atorvastatin was stirred in a minimum amount of acetone and isopropanol just below boiling point for over 24h. The solution was filtered, water added as anti-solvent and the product is allowed to crystallize in air at room temperature [86].
Water
Non-stoichiometric Channel
Solvate was crystallized from a 2-propanol: water (8:2, v/v) mixture in air at room temperature [87].
Cyclohexane
Stoichiometric Lattice Inclusion
Solvate isolated by vapor diffusion of cyclohexane into a toluene solution of Dirithromycin [88].
Table 4: Cases Representing the Synthesis of Different Types of Solvate Systems.
After a solvate has been formed, one needs to properly characterize it. Once again, single crystal XRD is the all-telling technique for characterization. The data obtained will result in the molecular structure which can be used to establish the intermolecular bonding between components, whether a “true solvate” has been formed, if the solvent is located predominantly in the voids. To explore this method, an Esomeprazole Magnesium solvate, displayed in Figure 12 will be investigated. Esomeprazole Magnesium is a poorly soluble pharmaceutical salt with low stability. In this solvate, water is bonded in two distinct modes: strong dative-covalent bonding directly with Mg and in the voids of the crystal lattice. The former water molecules are bonded more powerfully than the latter. The 1-butanol molecules are also found in the channels and can be seen to form hydrogen bonds with water. This compound is determined to be a “true solvate” with respect to the water molecules, and butanol is found in nonstoichiometric amounts.
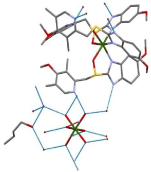
Figure 12: Esomeprazole Magnesium solvate with water and 1-butanol.
Hydrogen bonding between molecules is shown in blue [78].
Since single crystals are often not easily attainable, there are a variety of other techniques that can be used for characterization. PXRD is particularly useful when the solvate is synthesized via the formation of a new crystal lattice that incorporates the solvent. This will result in a PXRD pattern that is novel in comparison to the API [86]. Conversely, if the solvent is simply filling the voids in the crystal lattice, the PXRD pattern will appear to be analogous to that of the API with very slight differences. In this situation, it is necessary to conduct further analysis, such as ssNMR, to ensure that a solvate has indeed been formed [87]. An outstanding illustration of this is seen in Figure 13; 13CssNMR spectra are taken for a non-stoichiometric solvate where solvent is found in the channels. The solvate is then heated to remove solvent and the resulting spectra are observed, while PXRD patterns showed no differences as the solvent was removed [88].
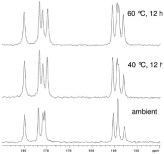
Figure 13: 13C ssNMR of a non-stoichiometric, channel solvate LY297802
tartrate at (a) ambient conditions, (b) elevated temperature with solvent
removal, and (c) elevated temperature with increased solvent removal [92].
Dynamic Vapor Sorption (DVS) is another valuable tool in analyzing pharmaceutical solvates. This instrument measures the mass of a sample with respect to changes in Relative Humidity (RH). For example, if a hydrate-forming API is put into an environment of increasing humidity, a change in mass is seen during hydrate formation. The additional mass will correspond to the ratio of water incorporated into the system, and the graph will form a hysteresis such as the one shown in Figure 14 [89]. This technique can also be used with other organic vapors to determine solvate formation [90]. Solvates that can form in multiple ratios or non-stoichiometrically will show multiple hysteresis. If solvate is found to bind only to the surface, it is easily removed and usually appears as a linear increase in mass with increasing RH [91]. Solvent that is incorporated into the crystal lattice will not instantly be removed; this is illustrated in Figure 14, as well. Once the solvate has formed, it takes a significant decrease in humidity to remove it from the lattice.
Solution NMR is not a useful technique to determine the amount of solvent present due to contaminations. Often the solvate is a hydrate, and water is found in many deuterated solvents [92]. A more appropriate method is to use thermal analysis such as DSC and TGA (thermogravimetric analysis). DSC will show the removal of solvent as an endothermic peak. Due to intermolecular interactions, solvent will evaporate above its boiling point. Channel solvent is usually removed much easier and closer to boiling temperatures. TGA monitors the weight loss as a substance is heated. The percentage lost (assuming the API has not decomposed) will correspond to how much solvent has been removed, which can then be used to calculate the ratio of sovent: API [93].
Excipient Selection and Solid-state Screening
There are a variety of methods to improve the solubility and dissolution rate of APIs; polymorphic screening, salt formation, solvate formation, eutectics formation, co-amorphization, co-crystal formation with an excipient, and co-crystal formation with a second API. In this section, the emphasis is based on the excipient selection. The excipient that is chosen for inclusion into a drug’s solid-state should increase the solubility and consequently the bioavailability of the drug. This is mainly done by choosing molecules that are watersoluble and have plenty of potential for intermolecular bonding [94]. It is also ideal to pick an excipient with a melting point lower than the API for reasons previously mentioned. The principal approach used in choosing an appropriate molecule is the supramolecular synthon method, which evaluates the potential intermolecular bonds between separate constituents of the multi-component solid-state [95]. Illustrated in Figure 15 are both hetero- and homosynthons: one involving a bond that interacts with the same type of bond (e.g. carboxylic-carboxylic) and the latter being dissimilar bond types interacting with each other (e.g. carboxylic-amide). It was reported that heterosynthons are more energetically stable; however, homosynthons are still a feasible means of intermolecular bonding [96]. The proposed intermolecular bonding should be strong enough to displace the bonding of the single components with themselves in order to synthesize the aforementioned solid-states.
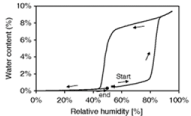
Figure 14: Hysteresis shown for an API with the capability to form a solvate.
Relative humidity is increased gradually and then reduced [89].
A database of approved excipients is easily accessible from the FDA [97], Table 5 is an example of the results for the molecule Ethylparaben (EPB). This excipient is pre-approved for use in drugs both orally and topically with potency varying depending on the method administered. If, for example, to form a co-crystal between an API and EPB, it should be in a ratio that will not cause EPB to be administered more than ~0.6mg per dose.
![]()
Inactive Ingredient
Route
Dosage Form
CAS Number
UNII
Maximum Potency
Ethylparaben
Oral
Granule, For Oral Solution
120478
14255EXE39
0.6mg
Ethylparaben
Oral
Powder, For Oral Solution
120478
14255EXE39
0.66mg
Ethylparaben
Oral
Powder, For Solution
120478
14255EXE39
0.6mg/2g
Ethylparaben
Oral
Solution
120478
14255EXE39
0.6mg/5 mL
Ethylparaben
Oral
Suspension
120478
14255EXE39
2mg/5 mL
Ethylparaben
Topical
Emulsion, Cream
120478
14255EXE39
NA
Table 5: Inactive Ingredient Search for Approved Drug Products, EPB.
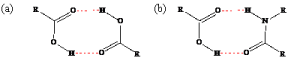
Figure 15: Hydrogen bonding motifs of (a) carboxylic-carboxylic homosython
and (b) a carboxylic-amide heterosynthon.
Where CAS is Chemical Abstracts Services, UNII is Unique Ingredient Identifier and EPB is ethylparaben.
Another source that researchers can use when choosing excipients is the Generally Regarded as Safe (GRAS) database, which is also published by the FDA [98]. Based on current studies, molecules are given an evaluation from 1-5, and the meanings of these ratings are specified in Table 6. Ascorbic acid, also known as vitamin C, is given a conclusion of 1 since it is well studied and known to be safe for consumption. Conclusions 1 and 2 are acceptable excipient choices for solid-state modifications. This database, however, can lead to some misleading results (e.g. nutmeg is listed as class 3, but it is ingested frequently in food). Also, since these inactive ingredients are not necessarily approved for use in drug products, new clinical testing may need to be conducted. The preferred method for excipient choice should be the Inactive Ingredient Search for Approved Drug Products Database.
![]()
Type of Conclusion
Definition
1
There is no evidence in the available information on [substance] that demonstrates, or suggests reasonable grounds to suspect, a hazard to the public when they are used at levels that are now current or might reasonably be expected in the future.
2
There is no evidence in the available information on [substance] that demonstrates a hazard to the public when it is used at levels that are now current and in the manner now practiced. However, it is not possible to determine, without additional data, whether a significant increase in consumption would constitute a dietary hazard.
3
While no evidence in the available information on [substance] demonstrates a hazard to the public when it is used at levels that are now current and in the manner now practiced, uncertainties exist requiring that additional studies be conducted.
4
The evidence on [substance] is insufficient to determine that the adverse effects reported are not deleterious to the public health should it be used at former levels and in the manner formerly practiced.
5
In view of the almost complete lack of biological studies, the Select Committee has insufficient data upon which to evaluate the safety of [substance] as a [intended use].
Table 6: Types of Conclusions from the Selection Committee on GRAS Substances.
Another source for finding excipients is the Everything Added to Food in the United States (EAFUS) lists [99]. As the name suggests, the list includes chemicals that the FDA has approved for additives in food. It is theorized that if the substance is safe for ingestion, then it can be used to some extent for oral administration.
The databases mentioned above are suitable to use when choosing an appropriate solvent for solvate formation; however, perhaps a more appropriate method is to use the ICH’s (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use) guideline for residual solvents. Solvents are given a Class 1-3 where Class 1 should be avoided for use in pharmaceutical production, 2 should be limited and 3 has low toxic potential. For solvate formation, one should only consider Class 3 solvents that have a permitted daily exposure of 50mg or more [100].
The molecule that is added for intermolecular interaction with the API may not always be an inactive ingredient. Recently, drugdrug compounds have become of increasing interest; they have been employed to reduce number of prescriptions, reposition drugs, reduce production cost and increase therapeutic effect [101]. It has been shown that combining antihypertension drugs that display poor aqueous solubility via co-crystal, co-amorphous mixture and eutectic formation leads to increased antihypertension activity [102]. As an outcome, number of prescriptions and production costs should decrease.
Now that an appropriate excipient has been chosen, which is arguably the most difficult task in this approach of crystal engineering, an intensive screening process must commence. While solid-state formation procedures are well studied, they are still not necessarily well understood, especially in terms of which combinations will form novel solid-states. Therefore, proper screening is essential for successful crystal engineering [103]. An extremely efficient means of screening involves grinding API and potential excipient together at varying molar ratios and collecting the DSC thermographs at a heating rate of 10K/min. This can only be done when there is no decomposition of the components before their melting [104]. When done without the presence of solvent (neat grinding), this technique is considered “green” and does not complicate the system with possible solvate formation such as with the LAG method [105]. If the resulting phase diagram gives a “w” shape, then co-crystal formation is possible; if a “v” shape is determined then there is likely a eutectic or co-amorphous product. If the melting endotherms are found not to converge, then the resulting product is merely a physical mixture, such as the illustration in Figure 16 [106].
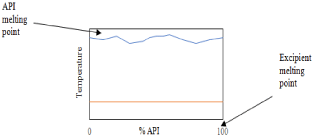
Figure 16: Binary phase diagram of an API and excipient incapable of
forming intermolecular interactions (physical mixture).
For a more rapid screening, one can merely run the DSC for ground mixtures of 1:1, 1:2 and 2:1 molar ratios of API: excipient [107]. One scenario is the presence of a melting endotherm that is distinct from the melting of the starting materials and is found at a consistent temperature in all three ratios. If the PXRD pattern shows novel peaks, then co-crystal formation is possible. Another scenario is the presence of an endothermic peak found lower than the melting temperature of the API or excipient that will alter slightly with varying molar ratio. Running PXRD will indicate if the product is co-amorphous or a eutectic.
While these techniques are both effective and reliable, they are not applicable for solvate formation, and sometimes solvent effects promote the formation of different solid-states (e.g. a co-crystal that can be synthesized from solution but not during the LAG process). Solution methods usually involve dissolving API (and often excipient) in solvent(s) and seeing what product is obtained via slow evaporation and/or cooling. Another technique is to create a suspension/slurry that is allowed to equilibrate at ambient conditions [108]. A clear drawback is that crystallization may occur slowly, and solubility issues could lead to single phases crystallizing out. Conceivably, screening with solvent(s) should be run in parallel to the DSC methods, so multiple solid-states can be screened simultaneously. Using solvent mixtures helps decrease the solubility differences between API and excipient and can promote co-crystal over solvate formation if low enough solvent concentrations are employed [109].
Another screening method, known as the Kofler technique, employs hot stage microscopy and is useful for solid-state screening. One component is melted and allowed to solidify, then a second molten phase is introduced. This partially solubilizes the first component in the second and a mixing zone is created that contains a concentration gradient (Figure 17). In this mixing zone, a novel solidstate may form under suitable conditions. Light microscope is used to observe the number of melting points while heating [110]. One melt is relevant for co-amorphous and eutectic mixtures, while two melts are seen for co-crystal. Co-crystal formation will result in a crystalline phase at the interface [111].
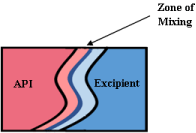
Figure 17: API (higher melting point) is melted and recrystallized before
excipient is brought into contact which creates a zone of mixing [110].
With appropriate screening and sufficient excipient/solvent selection, one can efficiently obtain a novel solid form that will potentially display increased solubility and bioavailability.
Conclusion
The FDA releases dozens of NMEs per year for pharmaceutical usage, and most of these drugs have very low aqueous solubility. The solubility of Class II and IV drugs can be improved through the crystal engineering technique of adding another molecule into the crystal lattice of the API. This molecule should be pre-approved for use in pharmaceuticals and not interact with the API. Use of this type of excipient requires no clinical testing, which is very advantageous to industry. Potential excipients can be narrowed down using a computerbased software that statistically analyzes molecular complementarity of the API with a list of common co-formers established from their molecular structure. Another technique, called the molecular synthon approach, will analyze the potential intermolecular bonding of the API and find corresponding excipients. Once a list of excipients is chosen, they should be screened using solvent crystallization in parallel with grinding and DSC techniques.
Should the screening result in the formation of a novel solidstate, it is essential that it is appropriately identified. The first step should be to have the proper characterization of both the API and the excipients used; this will lead to an easy comparison and solid-state analysis. Next, one should run PXRD on the new product. For the solid-states mentioned in this review (i.e. co-amorphous mixtures, co-crystals, eutectics and solvates), there are three different outcomes from the evaluation of the PXRD patterns. If the product’s pattern shows no new peaks and appears as a combination of the starting materials, then the result is either a physical mixture or a eutectic. If there are peaks that are distinct from the starting materials (and their polymorphs) or an entirely new pattern is observed, then the resulting solid-state could either be a co-crystal or solvate. The final option is to observe a “halo” in which there is simply one very broad peak. This pattern is characteristic of an amorphous material.
A good idea at this point would be to run 1H NMR on the product and compare it to that of the starting materials. Are the chemical shifts corresponding to the API present? How about the shifts for the excipient or solvent? Solution NMR is a valuable tool to ensure that the API did not decompose and determines what is present in their product. If a PXRD pattern of the product resulted in novel peaks and 1H NMR revealed that the excipient (or solvent) was present, then the result is most likely a co-crystal (or solvate). Likewise, if the PXRD pattern was amorphous and both starting materials are present in the NMR spectrum, then the product is co-amorphous. 1H NMR cannot be used to help elucidate physical mixtures from eutectics.
The next step would be to run thermal analysis to validate the formation of a new solid-state. If there is a single peak in DSC that is distinct from the melting of the starting materials, then the product is a co-crystal. If there is a mixture of co-crystal and starting material, then three peaks should be observed: one for eutectic melt, one for co-crystal melt and one for the melting of excess API. The melting point of the co-crystal is usually seen between the melting points of the starting materials. Solvates will often result in a thermograph that shows the evaporation of solvent in the lattice above its boiling point, followed by the melting of the API. TGA should be run on potential solvates to see if the weight loss corresponds to solvent loss. Coamorphous mixtures are not predictable in their DSC thermographs but will often not show the melting points of the starting materials, and an exothermic peak will be present. Physical mixtures will simply show the melting points of the starting materials in DSC, while eutectics will melt below the lower melting point starting material (usually excipient). DSC is also useful to plot phase diagrams. Eutectics and co-amorphous systems give a “v” shape, while co-crystals will result in a “w” shape. The melting points in physical mixtures will not converge, so no such shape will be present.
These techniques are usually sufficient in determining what solidstate is formed, but they can be aided by ssNMR, FT-IR and Raman. The ultimate tool would be to determine the molecular structure from single crystal XRD, but often it is difficult to grow crystals of appropriate quality. After a novel solid-state is formed, the solubility of this new entity should be determined and compared with that of the API. This is usually done by thermodynamic solubility tests and dissolution studies. If the novel product is found to have an increased solubility compared to the API, then a viable alternative to the API has been synthesized. The pharmaceutical company can now investigate patenting and eventually marketing this form.
References
- Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Järvinen T, et al. Prodrugs: design and clinical applications. Nat. Rev. Drug Discov. 2008; 7: 255-270.
- US FDA. The Biopharmaceutics Classification System (BCS) Guidance.
- Lindenberg M, Kopp S, Dressman JB. Classification of orally administered drugs on the World Health Organization Model list of Essential Medicines according to the biopharmaceutics classification system. Eur. J. Pharm. Biopharm.2004; 58: 265-278.
- Di L, Kerns EH, Carter GT. Drug-like property concepts in pharmaceutical design. Curr. Pharm. Des. 2009; 15: 2184-2194.
- Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 2000; 44: 235-249.
- Di L, Fish PV, Mano T. Bridging solubility between drug discovery and development. Drug Discov. Today. 2012; 17: 486-495.
- Zhou JL, Zhang ZL, Banks E, Grover D, Jiang JQ. Pharmaceutical residues in wastewater treatment works effluents and their impact on receiving river water. J. Hazard. Mater. 2009; 166: 655-661.
- Desiraju GR, Gautam R. Crystal engineering : the design of organic solids. Elsevier. 1989.
- Savjani KT, Gajjar AK, Savjani JK. Drug solubility: importance and enhancement techniques. ISRN Pharm. 2012; 195727.
- Yu L. Amorphous pharmaceutical solids: preparation, characterization and stabilization. Adv. Drug Deliv. Rev.2001;48: 27-42.
- Shah N, Sandhu H, Duk Soon C, Chokshi H, Malick W. Amorphous Solid Dispersions : Theory and Practice.
- Hancock BC, Parks M. What is the true solubility advantage for amorphous pharmaceuticals?. Pharm. Res. 2000; 17: 397-404.
- Yamamura S, Gotoh H, Sakamoto Y, Momose Y. Physicochemical properties of amorphous precipitates of cimetidine-indomethacin binary system. Eur. J. Pharm. Biopharm. 2000; 49: 259-265.
- Laitinen R, Löbmann K, Strachan CJ, Grohganz H, Rades T. Emerging trends in the stabilization of amorphous drugs. Int. J. Pharm. 2013; 453: 65-79.
- Kasten G, Grohganz H, Rades T, Löbmann K. Development of a screening method for co-amorphous formulations of drugs and amino acids. Eur. J. Pharm. Sci. 2016; 95: 28-35.
- Bates S, Zografi G, Engers D, Morris K, Crowley K, Newman A. Analysis of Amorphous and Nanocrystalline Solids from Their X-Ray Diffraction Patterns. Pharm. Res.2006; 23: 2333-2349.
- Keen DA, Dove MT. Mineral. Total scattering studies of silica polymorphs: similarities in glass and disordered crystalline local structure. Mag. 2000; 64: 447-457.
- Dengale SJ, Grohganz H, Rades T, Löbmann K. Recent advances in coamorphous drug formulations.Adv. Drug Deliv. Rev. 2016; 100: 116-125.
- Suresh K, Mannava MKC, Nangia A. A novel curcumin-artemisinin coamorphous solid: physical properties and pharmacokinetic profile. RSC Adv. 2014; 4: 58357-58361.
- Han Y, Pan Y, Lv J, Guo W, Wang J. Powder grinding preparation of co-amorphous β-azelnidipine and maleic acid combination: Molecular interactions and physicochemical properties. Powder Technol. 2016; 291: 110-120.
- Lim AW, Löbmann K, Grohganz H, Rades T, Chieng NJ. Investigation of physical properties and stability of indomethacin-cimetidine and naproxen-cimetidine co-amorphous systems prepared by quench cooling, coprecipitation and ball milling. Pharm. Pharmacol. 2016; 68: 36-45.
- Lefort R, De Gusseme A, Willart JF, Danède F, Descamps M. Solid state NMR and DSC methods for quantifying the amorphous content in solid dosage forms: an application to ball-milling of trehalose. Int. J. Pharm. 2004; 280: 209-219.
- Schantz S, Hoppu P, Juppo AM. A solid-state NMR study of phase structure, molecular interactions, and mobility in blends of citric acid and paracetamol. J. Pharm. Sci.2009; 98: 1862-1870.
- Shah B, Kakumanu VK, Bansal AK. Analytical techniques for quantification of amorphous/crystalline phases in pharmaceutical solids. J. Pharm. Sci. 2006; 95: 1641-1665.
- Jensen KT, Larsen FH, Cornett C, Löbmann K, Grohganz H, Rades T. Formation Mechanism of Coamorphous Drug-Amino Acid Mixtures. Mol. Pharm. 2015; 12: 2484-2492.
- Yamamoto K, Kojima T, Karashima M, Ikeda Y. Physicochemical Evaluation and Developability Assessment of Co-amorphouses of Low Soluble Drugs and Comparison to the Co-crystals.Chem. Pharm. Bull. (Tokyo). 2016; 64: 1739-1746.
- Chieng N, Aaltonen J, Saville D, Rades T. Physical characterization and stability of amorphous indomethacin and ranitidine hydrochloride binary systems prepared by mechanical activation. Eur. J. Pharm. Biopharm. 2009; 71: 47-54.
- Allesø M, Chieng N, Rehder S, Rantanen J, Rades T, Aaltonen J. Enhanced dissolution rate and synchronized release of drugs in binary systems through formulation: Amorphous naproxen-cimetidine mixtures prepared by mechanical activation. J. Control. Release. 2009; 136: 45-53.
- Widmann G. DSC of amorphous materials. Thermochim. Acta. 1987; 112: 137-140.
- Löbmann K, Laitinen R, Grohganz H, Gordon KC, Strachan C, Rades T. Coamorphous Drug Systems: Enhanced Physical Stability and Dissolution Rate of Indomethacin and Naproxen. Mol. Pharm. 2011; 8: 1919-1928.
- Chieng N, Hubert M, Saville D, Rades T, Aaltonen J. Formation Kinetics and Stability of Carbamazepine-Nicotinamide Cocrystals Prepared by Mechanical Activation. Cryst. Growth Des. 2009; 9: 2377-2386.
- Grifasi F, Chierotti MR, Gaglioti K, Gobetto R, Maini L, Braga D, et al. Using Salt Cocrystals to Improve the Solubility of Niclosamide. Cryst. Growth Des. 2015; 15: 1939-1948.
- Vishweshwar P, McMahon JA, Bis JA, Zaworotko M. Pharmaceutical cocrystals. J. J. Pharm. Sci. 2006; 95: 499-516.
- FDA; CDER. 2013.
- Aitipamula S, Banerjee R, Bansal AK, Biradha K, Cheney ML, Choudhury AR, et al. Polymorphs, Salts, and Cocrystals: What’s in a Name?. J. Cryst. Growth Des. 2012; 12: 2147-2152.
- Etter MC. Hydrogen bonds as design elements in organic chemistry. J. Phys. Chem. 1991; 95: 4601-4610.
- Faábián L. Cambridge Structural Database Analysis of Molecular Complementarity in Cocrystals. Cryst. Growth Des. 2009; 9: 1436-1443.
- Shyam Karki, Tomislav Friščić, William Jones, Motherwell WDS. Screening for Pharmaceutical Cocrystal Hydrates via Neat and Liquid-Assisted Grinding. 2007; 4: 347-354.
- Sungyup J, Insil C, Won II K. Liquid-Assisted Grinding to Prepare a Cocrystal of Adefovir Dipivoxil Thermodynamically Less Stable than Its Neat Phase. Crystals. 2015; 5: 583-591.
- Shewale S, Shete AS, Doijad RC, Kadam SS, Patil VA, Yadav AV. Formulation and Solid State Characterization of Nicotinamide-based Cocrystals of Fenofibrate. Indian J. Pharm. Sci. 2015; 77: 328-334.
- Sládková V, Cibulková J, Eigner V, *Šturc A, Kratochvíl B, Rohlíêek J. Application and Comparison of Cocrystallization Techniques on Trospium Chloride Cocrystals. Cryst. Growth Des. 2014; 14: 2931-2936.
- Zhang S. Physical Properties and Crystallization of Theophylline Co-crystals, KTH Chemical Science and Engineering, 2010.
- Hadi K. Spray Drying of Cocrystals for Engineering Particle Properties, Uppsala Universitet. 2015.
- Peterson ML, Stanton MK, Kelly RC, Staples R, Cheng A, Pople JA, et al. Preparation, solid state characterization, and single crystal structure analysis of N-(4-(6-(4-(trifluoromethyl)phenyl)pyrimidin-4-yloxy)benzo[d]thiazol-2-yl) acetamide crystal forms. CrystEngComm. 2011; 13: 1170-1180.
- Khorasani S, Fernandes MA, Hutter J, Levendis DC, Li NY, Abrahams BF. A synthetic co-crystal prepared by cooperative single-crystal-to-single-crystal solid-state Diels-Alder reaction. Chem. Commun. 2017; 3: 4969- 4972.
- Srirambhatla VK, Kraft A, Watt S, Powell AV. Crystal Design Approaches for the Synthesis of Paracetamol Co-Crystals. Cryst. Growth Des. 2012; 12: 4870-4879.
- Meng F, Li Y, Liu X, Li B, Wang L. Single-Crystal Structures and Typical Hydrogen-Bonding Motifs of Supramolecular Cocrystals Containing 1,4-Di(1H-imidazol-1-yl)benzene. Cryst. Growth Des. 2015; 15: 4518-4525.
- Lapidus SH, Stephens PW, Arora KK, Shattock TR, Zaworotko MJ. A Comparison of Cocrystal Structure Solutions from Powder and Single Crystal Techniques. Cryst. Growth Des. 2010; 10: 4630-4637.
- Mandala VS, Loewus SJ, Mehta MA. Monitoring Cocrystal Formation via In Situ Solid-State NMR. J. Phys. Chem. Lett. 2014; 5: 3340-3344.
- Tishmack PA, Bugay DE, Byrn SR. Solid-state nuclear magnetic resonance spectroscopy—pharmaceutical applications. J. Pharm. Sci. 2003; 92: 441- 474.
- Vogt FG, Clawson JS, Strohmeier M, Edwards AJ, Pham TN, Watson SA. Solid-State NMR Analysis of Organic Cocrystals and Complexes. Cryst. Growth Des. 2009; 9: 921-937.
- Lu J, Rohani S. Preparation and Characterization of Theophylline- Nicotinamide Cocrystal. Org. Process Res. Dev. 2009; 13: 1269-1275.
- Li P, Chu Y, Wang L, Wenslow RM, Yu K, Zhang H, et al. Structure determination of the theophylline-nicotinamide cocrystal: a combined powder XRD, 1D solid-state NMR, and theoretical calculation study. CrystEngComm. 2014; 16: 3141-3147.
- Padrela L, de Azevedo EG, Velaga SP. Powder X-ray diffraction method for the quantification of cocrystals in the crystallization mixture. Drug Dev. Ind. Pharm. 2012; 38: 923-929.
- Qiao N, Li M, Schlindwein W, Malek N, Davies A, Trappitt G. Pharmaceutical cocrystals: an overview. Int. J. Pharm. 2011; 419: 1-11.
- Srinivasulu Aitipamula K, Mapp LH, Wong AB, Shan Chow PH, Tan RB. Novel pharmaceutical cocrystals of triflusal: crystal engineering and physicochemical characterization. Cryst Eng Comm. 2015; 17: 9323-9335.
- Nayak A, Pedireddi VR. Pharmaceutical Cocrystals and Their Physicochemical Properties. Cryst. Growth Des. 2016; 16: 5966-5975.
- Yamashita H, Hirakura Y, Yuda M, Teramura T, Terada K. Detection of cocrystal formation based on binary phase diagrams using thermal analysis. Pharm. Res. 2013; 30: 70-80.
- Tilborg A, Leyssens T, Norberg B, Wouters J. Structural Study of Prolinium/ Fumaric Acid Zwitterionic Cocrystals: Focus on Hydrogen-Bonding Pattern Involving Zwitterionic (Ionic) Heterosynthons. Cryst. Growth Des. 2013; 13: 2373-2389.
- Nugrahani I, Bahari MUJ. The Dynamic Study of Cocrystal Formation between Anhydrous and Monohydrate Theophylline with Sodium Saccharine Dihydrate by FTIR. Chem Biochem. 2014; 2: 117-137.
- Elbagerma MA, Edwards MHG, Munshi T, Hargreaves M D, Matousek P, Scowen IJ. Characterization of new cocrystals by raman spectroscopy, powder X-ray diffraction, differential scanning calorimetry, and transmission raman spectroscopy. 2010; 10: 2360-2371.
- Lin HL, Hsu PC, Lin SY. Theophylline-citric acid co-crystals easily induced by DSC-FTIR microspectroscopy or different storage conditions. Asian J Pharm. Sci. 2013; 8: 19-27.
- Al-Saidan SM. J Control Release. Transdermal self-permeation enhancement of ibuprofen. 2004; 100: 199-209.
- Stoler E, Warner JC. Molecules. Non-Covalent Derivatives: Cocrystals and Eutectics. 2015; 20: 14833-14848.
- Law D, Wang W, Schmitt EA, Qiu Y, Krill SL, Fort JJ. Properties of rapidly dissolving eutectic mixtures of poly (ethylene glycol) and fenofibrate: The eutectic microstructure. J Pharm. Sci. 2003; 92: 505-515.
- Cherukuvada S, Nangia A. Eutectics as improved pharmaceutical materials: design, properties and characterization. Chem. Commun. 2014; 50: 906- 923.
- Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm. Biopharm. 2000; 50: 47-60.
- Goud NR, Suresh K, Sanphui P, Nangia A. Fast dissolving eutectic compositions of curcumin. Int. J. Pharm. 2012; 439: 63-72.
- Bi M, Hwang SJ, Morris KR. Thermochim. Acta. 2003; 404: 213-226.
- Jain H, Khomane KS, Bansal AK. Design and understanding of solid-state and crystalline materials. CrystEngComm. 2014; 16: 8471.
- Cherukuvada S, Guru Row TN, Cryst. Comprehending the Formation of Eutectics and Cocrystals in Terms of Design and Their Structural Interrelationships. Growth Des. 2014; 14: 4187-4198.
- Campanella L, Micieli V, Tomassetti M, Vecchio SJ. Them. Anal. Calorim. 2010; 99: 887-892.
- Fathy M, Hassan MA, Mohamed FA. Differential scanning calorimetry to investigate the compatibility of ciprofloxacin hydrochloride with excipients. Pharmazie. 2002; 57: 825-828.
- Figueirêdo CBM, Nadvorny D, de Medeiros Vieira ACQ, Soares Sobrinho JL, Rolim Neto PJ, et al. Enhancement of Dissolution Rate Through Eutectic Mixture and Solid Solution of Posaconazole and Benznidazole. Int. J. Pharm. 2017; 525: 32-42.
- Griesser UJ. The importance of solvates. In Polymorphism; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, FRG. 2006; 211-233.
- Khankari RK, Grant DJW. Pharmaceutical hydrates. Thermochim. Acta. 1995; 248: 61-79.
- Skieneh J, Khalili Najafabadi B, Horne S, Rohani S. Crystallization of Esomeprazole Magnesium Water/Butanol Solvate. Molecules. 2016; 21: 1-10.
- Grothe E, Meekes H, Vlieg E, ter Horst JH, de Gelder R, Parkin SRet al. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2011; 67: 324-328.
- Brittan HG. Polymorphism in Pharmaceutical Solids, Second Edition; CRC Press. 2009.
- Berzins A, Skarbulis E, Rekis T, Actins A. Crystal growth and design. Cryst. Growth Des. 2014; 14: 2654-2664.
- Nerenberg PS, Jo B, So C, Tripathy A. Head-Gordon, T. Optimizing Solute- Water van der Waals Interactions To Reproduce Solvation Free Energies. J. Phys. Chem. B. 2012; 116: 4524-4534.
- Mukherjee A, Tothadi S, Desiraju GR, Bauer JF. J. Valid. Technol. 2009; 15: 49-56.
- Chadha R, Kuhad A, Arora P, Kishor S. Characterisation and evaluation of pharmaceutical solvates of Atorvastatin calcium by thermoanalytical and spectroscopic studies. Chem. Cent. J. 2012; 6: 114.
- Tieger E, Kiss V, Pokol G, Finta Z, Dušek M, Rohlíček J, et al. CrystEngComm. 2016; 18: 3819.
- Stephenson GA, Stowell JC, Toma PH, Dorman DE, Greene JR, Byrn SR. Solid-State Analysis of Polymorphic, Isomorphic, and Solvated Forms of Dirithromycin. J. Am. Chem. SOC. 1994; 116: 5766-5773.
- Aitipamula S, Chow PS, Tan RBH. The solvates of sulfamerazine: structural, thermochemical, and desolvation studies. CrystEngComm. 2012; 14: 691- 699.
- Othman A, Evans JSO, Evans IR, Harris RK, Hodgkinson P. J. Pharm. Sci. 2007; 96: 1380-1397.
- Hilfiker R, Blatter F, Raumer M. In Polymorphism; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, FRG. 2006; 1-19.
- Beckmann W. Crystallization: basic concepts and industrial applications; Wiley-VCH. 2013.
- Alexander T, Siepmann J. Modern pharmaceutics. Volume 1: Basic principles and systems; Informa Healthcare. USA. 2009.
- Templeton AC, Byrn SR, Haskell RJ, Prisinzano TE. Discovering and developing molecules with optimal drug-like properties. 2015.
- Fulmer GR, Miller MAJ, Sherden NH, Gottlieb HE, Nudelman A, Stoltz BM, et al. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. 2010; 29: 2176-2179.
- Neville GA, Becksteadlf HD, Cooney JD. Thermal analyses (TGA and DSC) of some spironolactone solvatesFresenius. J Anal Chem.1994; 349: 746- 750.
- Shaw LR, Irwin WJ, Grattan TJ, Conway BR. The Effect of Selected Water- Soluble Excipients on the Dissolution of Paracetamol and Ibuprofen. Drug Dev. Ind. Pharm. 2005; 31: 515-525.
- Desiraju GR. Angewandte Chemie International Edition in English. 1995; 34: 2311-2327.
- Adalder TK, Sankolli R, Dastidar P. Homo- or Heterosynthon? A Crystallographic Study on a Series of New Cocrystals Derived from Pyrazinecarboxamide and Various Carboxylic Acids Equipped with Additional Hydrogen Bonding Sites. Cryst. Growth Des. 2012; 12: 2533- 2542.
- FDA.
- SCOGS (Select Committee on GRAS Substances).
- Schultheiss N, Newman A. Pharmaceutical Cocrystals and Their Physicochemical Properties. Cryst. Growth Des. 2009; 9: 2950-2967.
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. 2016.
- Sekhon BS. Drug-drug co-crystals. Daru. 2012; 20: 45.
- Haneef J, Chadha R. AAPS PharmSciTech. 2017.
- Yadav AV, Shete AS, Dabke AP, Kulkarni PV, Sakhare SS. Co-Crystals: A Novel Approach to Modify Physicochemical Properties of Active Pharmaceutical Ingredients. Indian J. Pharm. Sci. 2009; 71: 359-370.
- Berry DJ, Steed JW. Pharmaceutical cocrystals, salts and multicomponent systems; intermolecular interactions and property based design. Adv. Drug Deliv. Rev. 2017.
- Bag PP, Kothur RR, Reddy MC. Tautomeric preference in polymorphs and pseudopolymorphs of succinylsulfathiazole: fast evaporation screening and thermal studies. Cryst Eng Comm. 2014; 16: 4706.
- de Oliveira GGG, Feitosa A, Loureiro K, Fernandes AR, Souto EB, Severino P. Compatibility study of paracetamol, chlorpheniramine maleate and phenylephrine hydrochloride in physical mixtures. Saudi Pharm. J. SPJ Off. Publ. Saudi Pharm. Soc. 2017; 25: 99-103.
- Lu E, Rodríguez-Hornedo N, Suryanarayanan R. A rapid thermal method for cocrystal screening. Cryst Eng Comm. 2008; 10: 665.
- Zhang GGZ, Henry RF, Borchardt TB, Lou XJ. Efficient co-crystal screening using solution-mediated phase transformation. Pharm. Sci. 2007; 96: 990- 995.
- Rager T, Hilfiker R. Cocrystal Formation from Solvent Mixtures. Cryst. Growth Des. 2010; 10: 3237-3241.
- Berry DJ, Seaton CC, Clegg W, Harrington RW, Coles SJ, Horton PN, et al. Applying Hot-Stage Microscopy to Co-Crystal Screening: A Study of Nicotinamide with Seven Active Pharmaceutical Ingredients. Cryst. Growth Des. 2008; 8: 1697-1712.
- McNamara DP, Childs SL, Giordano J, Iarriccio A, Cassidy J, Shet MS, et al. Use of a glutaric acid cocrystal to improve oral bioavailability of a low solubility API. Pharm. Res. 2006; 23: 1888-1897.