
Abstract
Several mutations in MSTO1, encoding a mitochondrial fission factor, are associated with inherited muscular disorders, by affecting tissues and organs with high energy demands. Our infantile-onset case, slowly progressing, characterized by myopathy, cerebellar ataxia and dysarthria, has been followed at our Neuromuscular Center for delayed motor milestones, language and learning difficulty, scoliosis, arched palate and triangular face, but remained undiagnosed for thirty years. Whole-exome sequencing documented two causative variants (NM 018116:c.3_6del:p.Gly4Profs*68 and c.836G>A p.Arg279His) in MSTO1. The patient´s clinical presentation and imaging findings well overlap with the MSTO1 previously reported phenotype: early-onset muscle weakness, slowly progressive gait disturbances, elevated plasma creatine kinase (CK) levels, myopathic pattern, cerebellar and cognitive impairment, diffuse fatty degeneration at muscle MRI, and cerebellar hypotrophy in brain MRI. Taken together, these findings expand the genetic spectrum of this type of MSTO1 mutations, specifically affecting mitochondrial dynamics.
Keywords: MSTO1 myopathy; Waddling gate; Misato 1; Retinopathy
Introduction
MSTO1 gene, located on chromosome 1q22 (OMIM * 617619), encodes for Misato 1, a cytoplasmic protein that regulates mitochondrial morphology and distribution. Thanks to the diffusion of the next generation sequencing in the diagnostic practice, a growing number of pathogenic MSTO1 variants has been identified, associated with a complex phenotype, including myopathy, cerebellar ataxia, intellectual disability, dysmorfic features, pigmentary retinopathy and raised Creatine Kinase (CK) levels.
MSTO1-associated diseases are characterized by different age at onset, inheritance mode (AD, AR), skeletal muscle involvement (distal or proximal) and muscle histological features [1]. MSTO1 related myopathy is mostly recessive inherited and has mainly a congenital/infantile onset, with cerebellar and cognitive involvement and pigmentary retinopathy.
The first mutation in MSTO1 nuclear gene, was described in 2017 [2]. It is an extremely rare condition and to date mutations in MSTO1 have been documented in 28 additional patients with recessive transmission (VB) and in a single family with dominant trait 8VB.
Here we describe a novel MSTO1-related case in a 48-year-old woman followed since she was 3 years aged at our Neuromuscular Center, for delayed motor milestones, language and learning difficulty, muscle weakness, then associated with dysarthria and a mild progressive muscle and cerebellar involvement in the very late disease stages, harboring a compound heterozygosity for two MSTO1 pathogenic variants, detected by clinical exome sequencing: NM_018116:c.9_12del p.(Gly4Profs*68) in the first exon and NM_018116:c.836G>A p.(Arg279His) in exon 9.
Case Report
45-Years-Clinical History
Our proband was followed at our Neuromuscular Center, for delayed motor, language and learning milestones, starting from 1978. Her Caucasian parents were non-consanguineous, and family history was otherwise unremarkable. She started walking at 3 years of age, when scoliosis convex to the right, arched palate, triangular face, thick hair and arthrogryposis were first noted. Subsequently, she manifested a mild progressive muscular and cerebellar involvement, with dysarthria and predominant proximal hip girdle weakness, developing difficulties in jumping.
Initial diagnostic assessment included blood tests, muscular enzymes determination, neurophysiological studies, ECG 24-hour monitoring, echocardiography, respiratory function tests, brain and muscle MRI, muscle biopsy. Baseline blood testing resulted overall normal, including serum lactate levels, otherwise plasma Creatine Kinase (CK) was constantly elevated (min 218 - max 1750 UI/L).
A first muscle biopsy from the vastus lateralis muscle performed when she was 10 years old, displayed chronic myopathic changes: striking variability in muscle fibre size, fibre hypertrophy (data not shown). A second one, taken from the biceps brachialis muscle ten years later, showed the same findings, in particular simultaneous occurrence of central nucleus, and areas devoid of oxidative stains, as revealed by phosphorylase and oxidative enzyme staining (data not shown). A third one, taken from the deltoid when she was 45 aged, showed high fiber size variability, nuclear internalization and connective tissue increase. (Figure 1) Moreover, there was no histochemical signs of mitochondrial pathology.
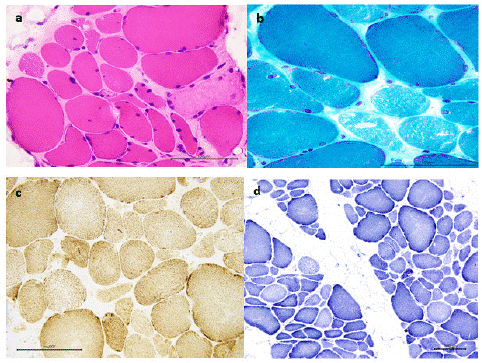
Figure 1: Third muscle biopsy (deltoid, 45 y.o.) showing high fiber size variability, nuclear internalization and connective tissue increase. Many fibers showing faint appearance and citoplasmic microvacuolization. Normal oxidative stains with minimal increase of subsarcolemmal mitochondria. A) Hematoxilin and Eosin (HE) staining; b) Gomori Trichrome (GT) staining; c) Succinate Dehydrogenase (SDH) and Cytochrome c Oxidase (COX) staining;
d) Nicotinamide Adenine Dinucleotide (NADH) tetrazolium reductase staining.
Brain MRI showed a marked early cerebellar hypotrophy; ECG 24-hour, echocardiogram and respiratory testing, regularly performed during follow up, were normal.
The first genetic testing performed included a 141-gene panel (Neuromuscular Genetics Panel) and dystrophin deletion/duplication sequencing analysis. None of the variants reported were considered relevant. The following genes were previously excluded by Sanger sequencing: ZNF9, MYOT, CAPN3.
Electromyogram (EMG) at 16 years resulted longstanding myopathic, nerve conduction and repetitive stimulation studies were normal, as described in previous studies [1].
A first muscle MRI, assessed when she was 39 years old, displayed overall a diffuse adipose involution in all the muscles of the pelvis, thighs, and legs, using turbo-spin echo T1-weighted sequences, except for vastus medialis, vastus intermedius and long abductor (Figure 2 a,b).
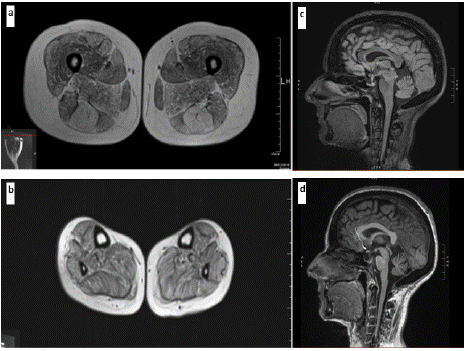
Figure 2: Muscle MRI images of the lower limbs (39 y.o.) documenting diffuse mild-to-moderate fat replacement of the a- thigs and b- legs, more evident in semitendinosus, rectus femoris and adductor magnus.
Brain MRI findings (43 y.o.): c- Fluid-Attenuated Inversion Recovery (FLAIR) sagittal showing evident cerebellar atrophy and d- T1- Weighted Imaging (T1WI) sagittal showing cerebellar atrophy.
A whole-body muscle MRI, performed four years after, documented hyperintensities on T1-weighted sequences in the torax, scapular, abdomen, pelvis region muscles, showing diffuse fatty infiltration, mostly in latissimus dorsi, anterior serratus, maximus gluteus, semitendinosus and sartorius muscles, while STIR positivity occurred in some muscles. (data not shown)
Disease slowly progressed over 40 years of follow-up and the patient's deambulation is actually still independent; now she is 48 years old and complains running loss, difficulty in postural changes and gait, mild proximal weakness, dysphagia, dysarthria, but these clinical findings remained static over the last years. A recent full neurological examination, especially targeted to a focused musculoskeletal assessment, has revealed only poor fine coordination, bilateral pes cavus, and some difficulties in raising her arms against downward pressure and over the head.
NGS Diagnosis
A recently performed targeted enriched NGS panel for myopathies and congenital myasthenic syndromes was negative, subsequently whole-exome sequencing (Illumina, TruSightOne) documented two variants in MSTO1 (NM_018116):
- a novel variant: c.9_12del p.(Gly4Profs*68) causing a premature stop codon;
- a previously reported missense change c.836G>A p.(Arg279His) [3]
Moreover, Sanger sequencing testing documented that her mother and the first brother were heterozygous for the missense variant (p.R279H), the second brother for the small deletion. Her father died before the genetic test (Figure 3 - pedigree)
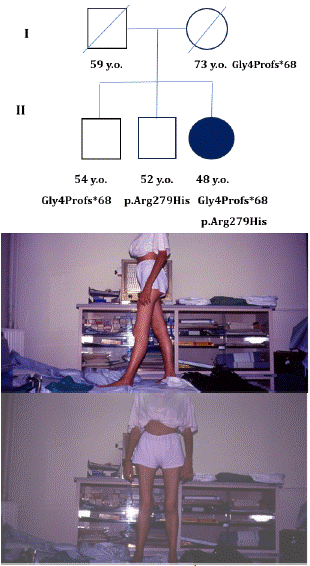
Figure 3: Clinical picture and family tree: a) genetic background of the proband and her relatives; b) clinical features of the patient at 20 y.o.: sagittal plane; c) clinical features of the patient at 20 y.o.: frontal plane.
In light of these findings, to best explore the effect of this MSTO1 pathogenic variants in our patient, more recently, we evaluated whether her phenotypic features overlapped with previously reported patients [4], including visual and cognitive impairment. Interestingly, a computerized Visual Field Test (c-VFT) showed a concentric loss of retinal sensitivity, but nor the ophtalmological examination revealed any abnormality of visual acuity, neither the visual evoked potential resulted altered, as reported in other studies [2].
An extended neuropsychological assessment evaluating intellectual, attentional, executive, social-cognitive, and language functioning was administered and she performed worse in the following tests: Addenbrooke's Cognitive Examination-Revised (ACE-R), Verbal Fluency Test, memory performance test, Rey Complex Figure Test (RCFT), showing a poor working memory and a global intellectual disability.
A recent brain MRI documented a mild progression of cerebellar atrophy and widening of subarachnoid spaces, suggestive of volume loss (Figure 2 c,d). Respiratory and cardiac assessment were overall normal.
Discussion
Mitochondrial diseases, that can be caused by mutations in either the mitochondrial DNA (mtDNA) or the nuclear DNA (nDNA), are phenotypically and genetically heterogeneous disorders which can affect mainly the most energy consuming tissues or organs, like the central nervous system, muscle tissues and the eyes, but the clinical manifestations are often non-specific [5].
To date, several mutations in genes encoding mithocondrial fission factor, such as the ubiquitously expressed MSTO1, are associated with inherited muscular disorders with multisystem features. MSTO1-related ataxia is an extremely rare disease among Caucasian populations [6]. Our infantile-onset case, mainly characterized by slowly progressing myopathy and cerebellar ataxia, unfortunately remained undiagnosed for 45 years. On the other hand, a follow-up for a longer time period is not reported overall in literature about MSTO1 myopathy until now.
The majority of patients with disease-associated variants in MSTO1 presents with biallelic variants suggesting autosomal recessive inheritance; however, one family has been reported with a single variant and presumed autosomal dominant inheritance. The pattern of inheritance we observed is consistent with the majority of previous reports suggesting an autosomal recessive disorder [1].
Thanks to NGS, variants were prioritized according to recessive inheritance and non-consanguinity of the parents. Thereby, she resulted compound heterozygous for two MSTO1 pathogenic variants: NM_018116:exon1:c.9_12del p.(Gly4Profs*68) in the first exon and NM_018116:c.836G>A p.(Arg279His) in exon 9, expanding both the definition of pathogenicity of some variants, and the assessment of genotype-phenotype correlations. Notably, our patient typically presented with delayed motor milestones and learning difficulties, but after 45-years-follow up she reported a very slow progression of the muscle weakness, and a mild progression of the cerebellar symptoms, like dysarthria, gait ataxia, dysmetria, and the cognitive impairment, without hearing or cardiac or respiratory involvement. Even the cerebellar atrophy at brain MRI resulted quite stable, as reported in other studies [7,8].
The p.(Arg279His) variant had been previously reported [8] in heterozygosity with a second pathogenic allele in three families. And this variant seems to be associated with a mild fatty infiltration of upper and lower leg muscles, as documented in MRI muscular imaging [3]; and it is also reported to cause pigmentary retinopathy, unlike in our patient. Bi-allelic loss-of-function MSTO1 variants can impair both the mitochondrial fusion and the mtDNA depletion. Despite its significance for mitochondrial dynamics, the compound heterozygous recessive mutations of MSTO1 are still unclear in humans until now. Nevertheless, our case suggests that the bi-allelic variants can cause an early-onset phenotype, characterized by a non-progressive muscular weakness and a stable congenital cerebellar atrophy.Therefore, it becomes increasingly important to have reliable natural history data to define genotype-phenotype correlation, outcome measures, and for potential therapeutic developments.
References
- Schultz-Rogers L, Ferrer A, Dsouza NR, Zimmermann MT, Smith BE, Klee EW, et al. Novel biallelic variants in MSTO1 associated with mitochondrial myopathy. Cold Spring Harb Mol Case Stud. 2019; 5: a004309.
- Nasca A, Scotton C, Zaharieva I, Neri M, Selvatici R, Magnusson OT, et al. Recessive mutations in MSTO1 cause mitochondrial dynamics impairment, leading to myopathy and ataxia. Hum Mutat. 2017; 38: 970-977.
- Donkervoort S, Sabouny R, Yun P, Gauquelin L, Chao KR, Hu Y, et al. MSTO1 mutations cause mtDNA depletion, manifesting as muscular dystrophy with cerebellar involvement. Acta Neuropathol. 2019; 138: 1013-1031.
- Chen J, Xiao J, Chen G, Xu Q, Wu X, Tian L, et al. Identification of novel MSTO1 compound heterozygous mutations in a Chinese family with recessive cerebellar atrophy and ataxia. Front Neurol. 2022; 13: 988519.
- Al Ojaimi M, Salah A, El-Hattab AW. Mitochondrial Fission and Fusion: Molecular Mechanisms, Biological Functions, and Related Disorders. Membranes (Basel). 2022; 12: 893.
- Li K, Jin R, Wu X. Whole-exome sequencing identifies rare compound heterozygous mutations in the MSTO1 gene associated with cerebellar ataxia and myopathy. Eur J Med Genet. 2020; 63: 103623.
- Ardicli D, Sarkozy A, Zaharieva I, Deshpande C, Bodi I, Siddiqui A, et al. A novel case of MSTO1 gene related congenital muscular dystrophy with progressive neurological involvement. Neuromuscul Disord. 2019; 29: 448-455.
- Iwama K, Takaori T, Fukushima A, Tohyama J, Ishiyama A, Ohba C, et al. Novel recessive mutations in MSTO1 cause cerebellar atrophy with pigmentary retinopathy. J Hum Genet. 2018; 63: 263-270.