
Editorial
Austin J Nutri Food Sci. 2014;2(1): 1008.
Inhibitory Properties of Kidney Bean Protein Hydrolysate and its Membrane Fractions Against Renin, Angiotensin Converting Enzyme, and Free Radicals
S. Mundi and Rotimi E. Aluko*
Department of Human Nutritional Sciences and the Richardson Centre for Functional Foods and Nutraceuticals, University of Manitoba, Winnipeg, Manitoba R3T 2N2, Canada
*Corresponding author: :Rotimi E. Aluko, Department of Human Nutritional Sciences, University of Manitoba, Winnipeg, MB R3T 2N2, Canada
Received: January 10, 2014; Accepted: February 10, 2014; Published: February 17, 2014
Abstract
Kidney bean hydrolysate (KBH) was obtained by alcalase hydrolysis of the seed globulin protein followed by membrane ultrafiltration to produce peptide fractions that differ in molecular sizes (<1, 1–3, 3–5, and 5–10 kDa). Evaluation of potential antihypertensive properties of the peptides showed that the <1 and 5–10 kDa fractions exhibited significantly highest (p<0.05) renin inhibition. In contrast, the KBH and peptide fractions showed similar and non–significant (p<0.05) inhibitory activities against angiotensin converting enzyme. The antioxidant power of the hydrolysates was evaluated through free radical scavenging activities (DPPH and hydroxyl radical), inhibition of iron activities (metal chelation and ferric reducing antioxidant power) and inhibition of linoleic acid peroxidation. The <1 and 5–10 kDa peptide fractions showed significantly (p<0.05) higher ability to scavenge DPPH free radical, inhibit peroxidation oflinoleic acid and reduce Fe3+ to Fe2+. Generally the fractions with <1 and 5–10 kDa peptides showed better potential as antihypertensive and antioxidant peptides, probably due to their slightly higher contents of hydrophobic aminoacids. It was concluded that kidney bean protein hydrolysate and some of the peptide fractions could potentially serve as useful ingredients to formulate functional foods and nutraceuticals against hypertension and oxidative stress.
Keywords: Kidney bean; protein hydrolysate; alcalase; renin; membrane ultrafiltration; angiotensin converting enzyme; antioxidant activity
Introduction
Several food proteins and peptides have been shown to display specific biological activities in addition to their proven nutritional value [1–3]. A growing body of scientific evidence reveals the positive impact of bioactive peptides and proteins on body function and human health by alleviating conditions such as coronary (ischemic) heart disease, stroke, hypertension, cancer, obesity, diabetes, and osteoporosis [4,5]. Specifically, research studies have shown some evidence for the efficiency of plant protein–derived peptides in improving hypertension or contributing to the overall antioxidant capacity of cells [6–9]. It has been reported that a large range of antihypertensive and antioxidant peptides and peptide mixtures (hydrolysates) have been produced from various food products such as beans, soy, corn, potato, peanut, milk, whey, egg, and meat proteins [3]. These peptides are inactive within the sequence of their parentproteins but can be released by chemical, enzymatic and microbial methods [3,10]. By far, the most effective and dependable method to produce peptides with the intended functionalities is enzymatic digestion [3]. Bioactive peptides are either produced in vivo by the action of gastrointestinal enzymes or obtained in vitro using specific enzymes, or during the preparation of certain foods. However, the source of proteins, the protein substrate pretreatment, the type ofenzymes used, and the hydrolysis conditions applied, all affect the efficacy of protein hydrolysates and type of peptides produced [3]. It is also known that the nature of residues in a peptide influences itsactivity. These peptides have the advantage of being naturally derived from food protein sources normally consumed as part of the daily diet, and they are considered to be milder and safer without the side effects associated with drugs. Peptides with antioxidant and ACE–inhibitory activities are usually rich in hydrophobic amino acids, which enhance absorption and interaction with target enzymes or free radicals [3,11].
High blood pressure confers a high risk of complications, as it is one of the major risk factors for cardiovascular diseases including coronary heart disease, peripheral artery disease and stroke [11,12]. Clinical evidence has shown that peptides released by the action of enzymes could be involved in the inhibition of the reninangiotensin– aldosterone system (RAAS), which is one key pathway for combating hypertension. In the RAAS, kidney–secreted renin cleaves angiotensinogen to produce an inactive decapeptide called angiotensin I; angiotensin converting enzyme (ACE) then removes a dipeptide from the C–terminal of angiotensin I to generate angiotensin II, a very potent vasoconstrictor that also enhances sodium (fluid) retention [11]. In addition, ACE is also responsible for inactivating the vasodilator bradykinin [13]. For this dual role in the maintenance of blood pressure and fluid and electrolyte homeostasis, inhibition of ACE has been successfully used for the treatment of hypertension and congestive heart failure. Synthetic ACE inhibitors such as captopril, enalapril, lisinopril and ramipril or the renin inhibitor (aliskiren) have been widely used for the effective clinical treatment of hypertension and heart failure in humans. However uses of these drugs are also associated with disadvantages, such as diarrhea, coughing, allergies,taste disturbances, skin rashes, impaired renal function, and in some cases excessively low blood pressure, i.e. hypotension [14–17]. For this reason, identification of possible natural sources of ACE inhibitors that have a strong antihypertensive activity and resistance to digestion by various proteases and with minimal negative side effect will be of great interest to formulators of functional foods. Although the effectiveness of the ACE–inhibitory activity may not be as high as those of synthetic drugs, many natural ACE–inhibitory peptides isolated from different food proteins could be applied in the prevention of hypertension and in the initial treatment of mildly hypertensive individuals [18].
Peptides have also been shown to be capable of inhibiting the uncontrolled oxidation of the biomacromolecules usually caused by reactive oxygen species (ROS). Peptides are known to act against an oxidative sequence by terminating chain reactions and removing free radical intermediates; therefore, they are able to reduce intensity of oxidative stress–related diseases like cancer, heart disease etc. Bioactive peptides block the oxidation process by neutralizing free radicals such as superoxide anion radical (O2−) and hydroxyl radical (.OH) which are products of regular metabolism [19]. Several in vitro studies have produced evidence that peptides generated from certain food proteins by enzymatic hydrolysis, including quinoa seed proteins [20], capelin protein [21], canola [22] and egg white protein [23] possess strong antioxidant activities. In particular, published studies have revealed strong evidence for the antioxidant activity for legume seed protein–derived peptides such as those from chickpeas [18,24– 26], cowpea [27], and soybean [28], but there is scanty information on the in vitro antioxidant and antihypertensive activity of kidney bean protein hydrolysate and the effect of peptide size on potency.
Kidney bean (Phaseolus vulgaris) is a pulse crop that contains high amount of proteins (20-30%) on a dry weight basis [29]. This puts them among some of the richest food sources of proteins, making them a good candidate to explore for the production of bioactive peptides. The goal of this study was to obtain an enzymatic protein hydrolysate from kidney bean globulin (major seed protein fraction) and fractionate the inherent peptides according to size using a membrane ultrafiltration system. The protein hydrolysate and membrane ultrafiltration fractions were then analyzed for in vitro antihypertensive (inhibition of ACE and renin) antioxidant (free radical scavenging, inhibition of linoleic acid oxidation, iron chelation, and ferric iron reducing power) activities.
Materials and Methods
Materials
Red kidney bean seeds were obtained from a local store in Winnipeg. Alcalase, N–(3–[2–furyl]acryloyl)–phenylalanylglycylglycine (FAPGG), glutathione (GSH), DPPH (2, 2–Diphenyl–1– picrylhydrazyl), 1–anilino–8–naphthalene sulfonate (ANS) and ACE (from rabbit lung) were purchased from Sigma–Aldrich (St. Louis, MO, USA). Human recombinant Renin Inhibitor Screening Assay Kit was purchased from Cayman Chemicals (Ann Arbor, MI, USA). Other analytical grade reagents and ultrafiltration membranes were obtained from Fisher Scientific (Oakville, ON, Canada).
Extraction and isolation of globulin protein
Red kidney bean seeds were ground into flour using a Retsch ZM200 centrifugal mill (Retsch GmbH, Haan, Germany). Globulinproteins were extracted from the flour according to the previously described ammonium sulfate precipitation method [30]. Briefly, an aqueous extract (obtained using 0.1 M phosphate buffer, pH 7.0 containing 0.4 M NaCl) of the flour was adjusted to 40% ammonium sulfate saturation, in order to precipitate smaller proteins and enzymes. After centrifugation (7000xg, 1 h, 4°C), the supernatant was then brought to 80% ammonium sulfate saturation to precipitate the globulins. The precipitating salt (ammonium sulfate) was then removed from the isolated globulins by dialyzing sample against water. The dialysis bag content was centrifuged (7000xg, 1 h, 4°C) and the resultant precipitate was freeze–dried as the globulin isolate.
Preparation and fractionation of kidney bean globulin protein hydrolysates
Proteolysis of the isolated kidney bean globulin isolate was conducted with alcalase. The globulin protein isolate (5%, w/v, protein weight basis) was suspended in deionized water in a reaction vessel equipped with a stirrer, heated to 50°C and adjusted to pH 9.0 prior to the addition of alcalase (4% w/w, based on the protein content of the protein isolate). The digestion was performed at the above stated conditions for 4 h with the pH of the reaction mixture maintained constant by addition of 2 M NaOH. At the end of the proteolysis period, the mixture was heated in boiling water for 10 min to inactivate alcalase; after cooling to room temperature, the mixture was adjusted to pH 4.0 with 2 M HCl to precipitate undigested proteins. Thereafter, the hydrolysate was centrifuged (30 min at 7,000xg). The supernatant containing target peptides was collected as the kidney bean protein hydrolysate (KBH) and a portion saved and stored at –20°C. The remaining liquid hydrolysate was passed through a 1 kDa membrane and the retentate passed through a 3 kDa ultrafiltration membrane. The retentate from 3 kDa membrane was passed through a 5 kDa whose retentate was then passed through a 10 kDa membrane. Permeates collected from each membrane were designated as <1, 1–3, 3–5 and 5–10 kDa peptide fractions, respectively. The KBH and membrane fractions were then freeze–dried and their protein contents determined by the modified Lowry method [31].
Amino acid analysis
An HPLC system was used for the analysis of the amino acid profiles after samples were hydrolyzed with 6 M HCl according to the method of Bidlingmeyer et al. [32]. The cysteine and methionine contents were determined after performic acid oxidation [33] while tryptophan content was determined after alkaline hydrolysis [34].
Surface hydrophobicity (So) determination
So of the KBH and the ultrafiltration peptide fractions was determined using a hydrophobic fluorescence probe, 1–anilino–8– naphthalene sulfonate (ANS) method as described by Hayakawa and Nakai [35] with some modifications. Samples were serially diluted to 0.0025–0.015% (w⁄v) in 0.01 M phosphate buffer (pH 7.0). Twenty µl of ANS (8.0 mM in 0.1 M phosphate buffer, pH 7.0) was added to 2 ml of sample solution. Fluorescence intensity of ANS–peptide conjugates was measured with an FP–6300 spectrofluorimeter (JASCO, Tokyo, Japan) at the excitation and emission wavelengths of 390 and 470 nm, respectively.
Determination renin–inhibitory activity
The method of Li and Aluko [6] was used to perform the renin inhibition assay using the Renin Inhibitor Screening Assay Kit. The background wells contained 20 µl of substrate, 160 µl of assay buffer, and 10 µl of Milli–Q. Thereafter, an aliquot of 20 µl of substrate, 150 µl of assay buffer, and 10 µl of Milli–Q water were added to the blank wells while 20 µl of substrate, 150 µl of assay buffer, and 10 µl of KBH or peptide fraction (final assay concentration of 1 mg protein⁄ml) were added to the inhibitor wells. The reaction was initiated by adding 10 µl of renin to the control and sample wells. The microplate was shaken for 10 s to mix, incubated at 37°C for 15 min, and then fluorescence intensity (FI) was recorded using an excitation wavelength of 340 nm and emission wavelength of 490 nm.
The percentage inhibition was calculated as: % Renin inhibition = FI (blank) – FI (sample) x 100⁄FI (blank)
Determination of ACE–inhibitory activity
The ACE–inhibitory activity was assayed as described by Udenigwe et al. [36] using FAPGG as substrate. Briefly, 1 ml of 0.5 mM FAPGG (dissolved in 50 mM Tris-HCl buffer containing 0.3 mM NaCl, pH 7.5) was mixed with 20 µl of ACE (1 U⁄ml; final activity of 20 mU) and 200 µl of KBH or peptide fractions (final assay concentrationof 1 mg protein⁄ml) in 50 mM Tris–HCl buffer. The decrease in absorbance at 340 nm, which is due to cleavage of the Phe–Gly peptide bond of FAPGG was recorded for 2 min at room temperature. For the blank experiment, Tris–HCl buffer was used instead of peptide fraction solutions. All experiments were performed in triplicate. Thepercentage inhibition of ACE was calculated as:
% ACE inhibition = Abs of blank–Abs of sample x 100⁄Abs of blank
DPPH radical scavenging assay
Reduction of DPPH by an antioxidant usually results in a loss of absorbance at 517 nm. The extent of discoloration of the solution indicates the scavenging efficiency of the added compound. Determination of antioxidant activity of KBH and the peptide fractions was adapted from the method described by Hou et al. [37] using a 96–well microplate. The KBH and the peptide fractions (final assay concentration of 1 mg protein⁄ml) were dissolved in 0.1 M sodium phosphate buffer, pH 7.0 containing 1% (w⁄v) Triton X–100. A solution of DPPH was prepared in methanol to a final concentration of 100 µM. Samples (100 µl) were added to 100 µl of DPPH in a 96– well microplate. A blank well contained only DPPH and the sodium phosphate buffer. The plate was then covered and incubated in the dark at room temperature for 30 min; absorbance of the sample (As)and blank (Ab) at 517 nm was measured in a spectrophotometer. The scavenging activities of KBH and the peptide fractions were compared to that of GSH. The percent scavenging activity of GSH and the samples was calculated using the following equation: DPPH Radical Scavenging Activity (%) = (Ab–As⁄Ab) x 100
Hydroxyl radical scavenging assay
The hydroxyl radical scavenging activity was measured according to the protocol previously described [38]. KBH, peptide fractions, GSH and 1, 10–phenanthroline (3 mM) were each separately dissolved in 0.1 M sodium phosphate buffer (pH 7.4) while FeSO4 (3 mM) and0.01% hydrogen peroxide were each separately dissolved in distilled water. An aliquot (50 µl) of 1, 10–phenanthroline and 50 µl of FeSO4 were added consecutively to 50 µl of KBH, peptide fractions, GSH, or buffer (control) in a clear, flat bottom 96–well microplate. Final assay concentration of samples was 1 mg protein⁄ml. To initiate reaction in the wells, 50 µl of hydrogen peroxide (H2O2) solution was added to the mixture, which was then covered and incubated at 37°C for 1 h with shaking. Thereafter, the absorbance of the mixtures was measured at 536 nm every 10 min for a period of 1 h. The absorbance was also determined for a blank (without peptides and H2O2) and a blank(without peptides). The ?OH scavenging activity was calculated as described by Ajibola et al. [38].
Determination of Fe2+ chelating activity
The iron chelating activity of KBH and peptide fractions was measured following the ferrozine method as described by Ajibola et al. [38]. KBH, peptide fraction or GSH solutions (final concentration of 1 mg protein⁄ml) was mixed with 0.05 ml of 2 mM FeCl2 and 1.85 ml distilled water in a reaction tube. Thereafter, 0.1 ml of 5 mM Ferrozine solution was added and mixed thoroughly. The mixture was allowed to stand at room temperature for 10 min followed by removal of 200 µl aliquot of the reaction mixture and added to a clear bottom 96–well plate. The control experiment contained all the reaction mixtures except that distilled water was used to replace the sample. Absorbance of sample (As) and blank (Ab) was measured using a spectrophotometer at 562 nm and the metal chelating activity of the sample was compared to that of GSH. The percentage chelating effect (%) was calculated using the following equation:
Fe2+ chelating activity (%) = (Ab–As⁄Ab) x 100
Ferric reducing antioxidant power (FRAP) assay
The ability of the hydrolysate to reduce iron (III) was determined according to the method of Yildirim et al. [39] with some modifications. Different concentrations of KBH, peptide fractions or GSH (1, 5 and 10 mg⁄ml) in 250 µl of distilled water were mixed with phosphate buffer (250 µl of 0.2 mM, pH 6.6) and 250 µl of 1% potassium ferricyanide solution dissolved in distilled water. The mixture was incubated at 50oC for 30 min, followed by addition of 250 µl of 10% (w⁄v) trichloroacetic acid. The mixture was then centrifuged at 1000xg for 10 min. Finally, 250 µl of the supernatant solution was mixed with 50 µl of distilled water and 50 µl of 0.1% (w⁄v) ferric chloride solution followed by addition of distilled water (200 µl). After 10 min reaction the absorbance of the resulting solution was measured at 700 nm. Increased absorbance of the reaction mixture indicated increased reducing power.
Inhibition of linoleic acid oxidation
Linoleic acid oxidation was measured using a previously described method [38]. KBH, peptide fractions or GSH were dissolved in 1.5 ml of 0.1 M phosphate buffer, pH 7.0 at a final concentration of 1 mg protein⁄ml. Each mixture was added to 1 ml of 50 mM ethanolic linoleic acid and stored in a glass test tube kept at 60°C in the dark for 7 days. On a daily basis, 100 µl of the reaction mixture was removed and mixed with 4.7 ml of 75% aqueous ethanol, 0.1 ml of ammonium thiocyanate (30%, w⁄v) and 0.1 ml of 0.02 M acidified ferrous chloride (dissolved in 1 M HCl). An aliquot (200 µl) of the resulting solution was added to a clear bottom 96–well microplate and the degree ofcolor development was measured using the spectrophotometer at 500 nm after 3 min incubation at room temperature.
Statistical Analysis
Data were collected in triplicates and subjected to one way analysis of variance using Statistical Analysis System Software (SAS version 9.2, SAS Institute, Cary, NC). Significant differences were determined by Duncan’s multiple range test and accepted at p<0.05.
Result and discussion
Protein hydrolysis
Enzymatic proteolysis of kidney bean globular protein and subsequent fractionation of the resultant KBH by membrane ultrafiltration resulted in fractions rich in small size (<10 kDa) peptides. The percent gross yield of KBH was 78%, and approximately 30.7, 20.3, 17 and 18% of peptides in the KBH had molecular weights of <1, 1–3, 3–5 and 5–10 kDa, respectively. The final retentate (>10 kDa fraction), which contained large size peptides had a yield of 14%. The protein contents were ˜87, 96, 89, 78 and 90% for the <1, 1–3, 3–5, 5–10 kDa and the KBH, respectively. The high yield of KBH reflects defficient digestion of the globular proteins by alcalase.
Amino acid analysis
Amino acid analysis of the unhydrolyzed and alcalase–treated globulins from kidney bean seed as well as the peptide fractions collected as permeates from 1, 3, 5 and 10 kDa membrane cut–offs are shown in Table 1. The amino acid analysis of the unhydrolyzed globulin meets the FAO’s 35% recommendation for essential amino acid content [40]. Protease hydrolysis of the globulin proteins affected the amino acid content of the fractions in various ways. Generally, theunhydrolyzed globulin and KBH as well as the peptide fractions all contained low levels of methionine and cysteine, which is typical of legume proteins. Conversely, all samples had high contents of glutamic acid, glutamine, aspartic acid, asparagine, lysine and alanine. Except for proline, cysteine, isoleucine valine and histidine which were relatively higher, most of the amino acid contents of the KBH were slightly lower than the unhydrolyzed globulin. In contrast, Pownall et al. [41] investigated the amino acid composition of pea peptides fractions separated from the <3 kDa permeate using high performance liquid chromatography (HPLC) and found an increase in hydrophobic (both aliphatic and aromatic) amino acid of the hydrolysate when compared with the isolate. In our study, when compared to the KBH, most peptide fractions showed higher average surface hydrophobicity. Pownall et al. [41] and Megías et al. [24] also reported higher contents of certain hydrophobic amino acids in relation to the original proteinhydrolysate. Hydrolysis markedly increased the cysteine content of the KBH and all the peptide fractions even though the other sulphur–containing amino acid, methionine was slightly reduced in all the fractions. The total hydrophobic aliphatic amino acid (valine, isoleucine and leucine) content showed the highest concentration in the <1 kDa fraction. The percentage of valine also increased in the hydrolysate and in the polypeptides with 5–10 kDa size, but was least in the 3–5 kDa fraction. The percentage content of leucine and isoleucine residues as well as hydrophobic aromatic amino acid (phenylanaline and tyrosine) were highest in the <1 kDa fraction, decreasing as the membrane MW cut–off increased from 1 kDa to 10 kDa. Proline, aslightly hydrophobic amino acid increased in the peptide fractions as MW cut–off was increased. Asparagine⁄aspartic acid and glutamine⁄ glutamic acid contents were also increased in the peptide fractions with exception of the 5–10 kDa fractions.
Surface hydrophobicity
Proteolysis of proteins may cause changes in protein globular structure as the hydrophobic regions hidden within the native protein are exposed [42]. After proteolysis, more hydrophobic amino acids are exposed because of the catalytic specificity of alcalase, which cleaves peptide bonds formed by these specific amino acid residues [43]. Quantitative structure–activity relationship studies of ACEinhibitory peptides have shown that peptides composed of amino acids with strongly hydrophobic (or aromatic) side chains have potent ACE–inhibitory activities [44,45]. Hydrophobicity is also a very important contributing factor to the activity of antioxidative peptides. This is because, the surface hydrophobic site is partly responsible for formation and maintenance of the spatial structures as well as protein interactions, including binding to cell membranes, protein-protein recognition, and formation of complexes with biologically active compounds [46]. Since the hydrophobic interactions are the driving forces for manifestation of the physiological functions of peptides, information on the hydrophobic character of bioactive peptides could contribute to further understanding of their mechanism of action. To determine hydrophobicity of the peptides, a hydrophobic fluorescent dye (ANS) was used as a probe. As shown in Figure 1, the affinity of ANS for hydrophobic patches increased from the <1 kDa fraction to the 5–10 kDa. This is probably because there are more oligopeptides available as the size of the membranes increased with larger surface containing the exposed hydrophobic residues. Wu et al. [47] also observed a decrease in the surface hydrophobicity of smaller soypeptides prepared by longer time papain hydrolysis compared to the larger peptides produced by shorter time hydrolysis. The authors attributed the observed difference to fewer hydrophobic binding sites on the smaller peptides compared to the larger peptides. The same authors also observed lower surface hydrophobicity for the ultrafiltrates than the hydrolysates [47]. On a similar note Molina Ortiz et al. [48] also observed that smaller chain peptides had less surface hydrophobicity. However, our results differ from those reported by Wang and co–workers [49] for papain hydrolysates of wheat gluten who showed that the permeate with a molecular weight cut–off of 5 kDa had higher surface hydrophobicity than the hydrolysate. The trend for the surface hydrophobicity is not consistent with the hydrophobic amino acid residue content, which suggests that arrangement of the amino acids differ between the peptide chains.
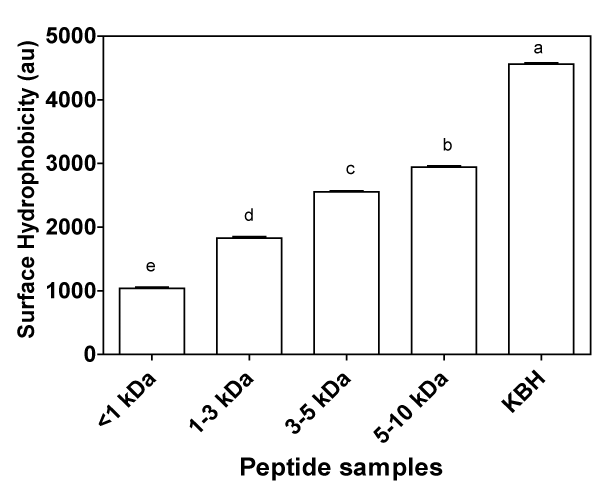
Figure 1: Surface hydrophobicity of kidney bean protein hydrolysate (KBH) and peptide fractions (<1,1–3,3–5,&5–10 kDa) in 10 mM phosphate buffer (pH 7.0). Bars with different alphabets have mean values that are significantly (p<0.05) different.
Renin– and ACE–inhibitory activities
Stable blood pressure is usually maintained by a balance between the plasma levels of the hypertensive peptide, angiotensin II, andthe hypotensive peptide, bradykinin [50]. This balance is highly dependent on renin and ACE; excessive activities increase level of angiotensin II and reduce bradykinin levels, hence increase in blood pressure. Therefore, peptides that inhibit activities of either or both enzymes could help restore normal blood pressure. Ultrafiltration separation of the KBH led to increased renin–inhibitory activities of the peptide fractions as shown in Figure 2A. Renin–inhibitory activity is related mostly to the type of amino acids present within the inhibitory peptide chains [51]. As indicated by the amino acid analysis (Table 1), peptide fractions with molecular masses less than 1 kDa and 5–10 kDa have higher contents of hydrophobic (valine, isoleucine and leucine) and aromatic amino acids (phenylalanine and tryptophan). Therefore, the higher renin–inhibitory activities of the <1 kDa and 5–10 kDa peptide fractions may be attributed to the increased levels of hydrophobic and aromatic amino acids when compared to the 1–3 and 3–5 kDa peptide fractions. In a previous work on peptide–like compounds, renin–inhibitory capacity values that are similar to those of the <1 kDa and 5–10 kDa peptide fractions have also been reported [52]. Similar to the report of Udenigwe et al. [51], it is possible that the nature and position of the amino acid rather than size of peptides play a major role in enhancing renin inhibition.
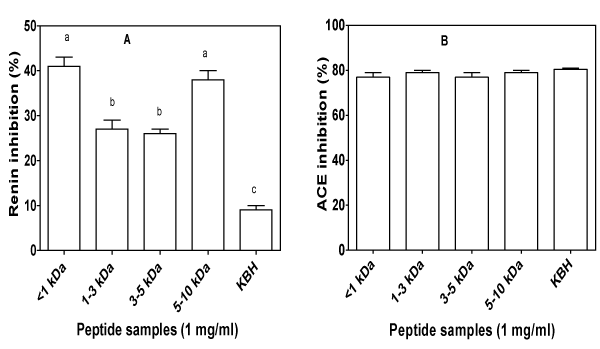
Figure 2: In vitro inhibitory activity of kidney bean hydrolysate (KBH) and peptide fractions (<1,1–3,3–5,&5–10 kDa) against renin (A) and angiotensin converting enzyme (B). For each plot, bars with different alphabets have mean values that are significantly (p<0.05) different.
ACE–inhibitory peptides that have shown very important role in regulating blood pressure through the direct action of angiotensin II on blood vessels range from dipeptides to oligopeptides [50]. In the current study, the ability of KBH and peptide fractions to inhibit in vitro activity of ACE is shown in Figure 2B. All peptide fractions and the KBH showed high percentage (above 77%) of ACE–inhibitory activity. The highest ACE inhibition of 80% was shown by KBH while <1 and 3–5 kDa fractions showed the minimum inhibition at 77%: however, there was no significant (p>0.05) difference between the ACEinhibitory activities of all the samples. The peptide fractions and KBH showed higher ACE–inhibitory activity than mung bean hydrolysate prepared with neutrase [53]. The differences in these findings may be explained by differences in the enzyme type, the concentration of the sample used, as well as the enzyme–to–substrate ratio, all of whichcould affect the type of peptides produced. Compared to the current study, Valdez–Ortiz et al. [54] reported higher inhibitory activity (90– 99%) for alcalase and thermolysin treated Azufrado (sulphur yellow) beans. In contrast to the ACE–inhibitory activity reported for hardto– cook black bean protein hydrolysate fractions [55], which was significantly dependent on peptide fraction molecular weight, the inhibitory activity in the present study had no particular trend.
Free radical scavenging properties
The free radical scavenging activity of KBH and peptide fractions was tested by measuring their ability to quench DPPH and hydroxyl radicals (Figure 3). The DPPH radical is reduced by conversion to a colourless product in the presence of antioxidants that possess hydrogen–donating or chain–breaking properties [56]. DPPH radicalscavenging activities of different fractions and the hydrolysate are shown in Figure 3A. Peptide fractions, but not hydrolysates possessed moderate to mild DPPH scavenging activities. The results varied depending on the peptide size with the <1 kDa and 5–10 kDa showing13 and 14%, which is less than 50% of the 32% scavenging capacity obtained for the positive standard (GSH). The DPPH inhibitory activities of 1–3 and 3–5 kDa fractions were significantly lower (p<0.05) than activities of <1 and 5–10 kDa fractions. Unlike the study by Girgih et al. [57] who reported that the DPPH inhibitory activity of hemp peptides depended on the molecular size, the observed pattern of DPPH–scavenging in the current study showed no relationship with molecular size, because activity significantly decreased (p<0.05) from 1 kDa to 1–3 kDa but was increased only slightly for the 3–5 kDa fraction and significantly for the 5–10 kDa fraction. The KBH which is a mixture of peptides including <1 to the 5–10 kDa sizes did not demonstrate any ability to scavenge free radical, possibly because of dilution effect, i.e. the ratio of inactive peptides was higher than that of active peptides. Thus, the ultrafiltration–mediated separation led to reduced ratio of the inactive peptides in the peptide fractions. In general, the peptide fractions from KBH were able to act as a DPPH radical scavengers probably because of their electrondonatingcapacity, and as result, form stable radicals or nonreactive conjugates through radical–peptide condensation [3]. The presence of certain amino acid residues, notably histidine, tyrosine, tryptophan, methionine, cysteine, and proline, amongst other aromatic and hydrophobic amino acids, have been reported to be associated with radical quenching activity of peptides [10]. The higher DPPH inhibitory activity shown in this study for <1 and 5–10 kDa peptide fractions could have been due to higher contents of hydrophobic aliphatic (valine, isoleucine and leucine) and hydrophobic aromatic (phenylanaline and tyrosine) amino acid residues in the two fractions, when compared to the 1–3 and 3–5 kDa fractions. The DPPH radical scavenging activity of KBH was lower than 48–58% for barley hydrolysate at 0.5 mg⁄mL [58] and 43–78% at 1.5 mg⁄ml assay concentration of smooth hound (Mustelus mustelus) muscle protein hydrolysates [59].
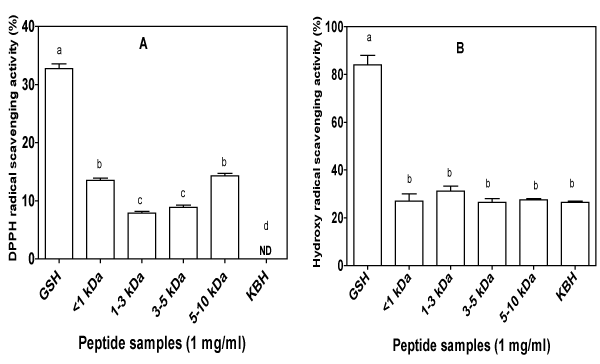
Figure 3: DPPH (A) and hydroxyl radical (B) scavenging activities of kidney bean protein hydrolysate (KBH) and peptide fractions (<1,1–3,3–5,&5–10 kDa) with glutathione (GSH) as a positive control. ND: no detectable activity. For each plot, bars with different alphabets have mean values that are significantly (p<0.05) different.
Figure 3B shows the hydroxyl radical scavenging properties of KBH and membrane fractions. It has been shown that protracted and severe oxidative stress in humans can lead to the initiation or promotion of a large number of chronic disease conditions [60]. Superoxide and hydrogen peroxide conversion as well as metalcatalyzed processes generate the highly reactive hydroxyl radical (•OH) which can oxidize virtually all organic cell constituents including DNA, lipids and proteins [60]. Once created, the •OH starts a chain of damages by reacting with target molecules that are vital for normal functioning of the cell, or which can become activated and pass the damage to vital cell components. Protein enzymatic hydrolysates and fractions obtained by using various molecular weight cut–off membranes may contain antioxidant peptides which rendered their protective actions in scavenging radicals. As evident in Figure 3B, the KBH and peptide fractions showed similar levels of •OH scavenging activity with no significant difference (p>0.05). Based on this result, it does not appear that the observed antioxidant activities were impacted by the hydrophobic residues in the hydrolysate and fractions presented. An antioxidant that works against •OH probablyacts by blocking formation of the radical from precursors such as superoxide and hydrogen peroxide or by chelating the transition metal ions such as Fe(II). Yen and Hsieh [61] attributed the hydroxyl radical scavenging potential to the combined effects of reducing power, donation of hydrogen atoms and scavenging of active oxygen. However in the current study, •OH scavenging ability of the KBH and its peptide fractions was not consistent with their ability to reduce Fe (III) to Fe (II) or to scavenge DPPH radical.
Iron chelating activity and FRAP
Studies with rodents [62,63], complemented by post–mortem human brain tissue analyses [64] have shown that aging is accompanied by a rise in the levels of iron and other metals (e.g., copper, zinc) in the biological system. Iron has been found to be involved in several key pathogenic processes in a number of degenerative diseases especially in neurodegenaration. For instance, the redox–active Fe (II) reacts with H2O2 to generate the highly reactive •OH (hydroxyl radical) via the Fenton reaction. High levels of •OH may lead to the development of various oxidant–induced metabolic disorders. Thus, there is a critical link between Fe(II) concentration and oxidative stress in the human biological environment. The crucial role of iron in promoting oxidative stress suggests that chelating agents such as bioactive peptides that can sequester free redox–active iron from its sites of activity to form nontoxic metal complexes could play an important role in preventing oxidative injury. The capacity of the KBH and the fractions was assessed for their ability to compete with ferrozine for ferrous ions, resulting in reduced absorbance of the ferrozine–Fe (II) complex as shown in Figure 4A. As expected, GSH showed the significantly highest (p<0.05) iron–chelating activity. The samples exhibited iron–chelating potency to various extents, with fractions containing peptides with the 3–5 and 5–10 kDa sizes showing significantly higher (p<0.05) chelating activity when compared to the KBH and 1–3 kDa fraction. This report is different from that of Girgih et al. [57] who reported the highest iron chelating activity for unfractionated hemp seed hydrolysate, attributing the effect to additive peptide effects. Samples that have shown some potency toscavenge Fe(II) can also scavenge other bivalent transition metal ions (e.g. Zn2+ and Cu2+), and hence would partially inhibit the propagation of lipid peroxidation that forms reactive oxygen radicals. This is because stabilization of metal prooxidants through sequestering inhibits free radical production. In addition, when peptides bind metal ions, the redox cycling capacity which is important for some metal–catalyzed oxidation may change. For instance, in ferritin the less reactive ferric ion (Fe3+) is localized in the polypeptide’s cavity, where it nucleates and aggregates to form ferric hydroxide core unable to be converted to the more reactive ferrous (Fe2+) specie [65]. The disruption of iron redox equilibrium leads to a reduction of free Fe2+, thereby preventing the decomposition of hydroperoxides [3]. Peptides with amino acid residues containing phosphorylated hydroxyl side chain groups (serine and threonine) and carboxyl groups (glutamic acid and aspartic acid) are good metal–ion binders [3]. Therefore, the high metal chelating activities of 3–5 and 5–10 kDa fractions may be due to their high contents of glutamic acid, aspartic acid, threonine and histidine. It is probable that hydrolysis of peptide bonds led to enhanced Fe2+ binding due to increased concentration of carboxylic and amine groups in acidic and basic amino acids [38]. Histidine residues have also shown efficiency in metal chelation because of its imidazole ring [38,66]. In fact, the strong Fe2+ and Cu2+ binding power of the endogenous dipeptide carnosine in muscle tissue has been attributed to the histidine residue [3]. Over all, the KBH and its ultrafiltrates showed reduced effectiveness as iron chelator when compared with Jamapa phaseolin hydrolysates (81%) [67] and the 97% iron chelating activity that was previously described for pea protein hydrolysates after treatment with thermolysin [41].
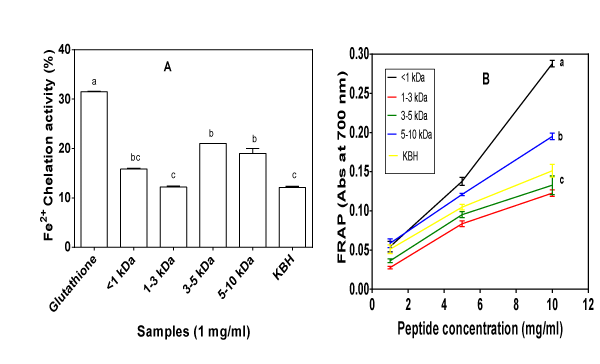
Figure 4: A. Percentage iron chelation ability and B. Dose dependent ferric reducing antioxidant property (FRAP) of kidney bean protein hydrolysate (KBH) and peptide fractions (<1,1–3,3–5,&5–10 kDa). For each plot, bars or lines with different alphabets have mean values that are significantly (p<0.05) different.
Results of the FRAP assay for KBH and peptide fractions are shown in Figure 4B. The reducing power assay is often used to evaluate ability of an antioxidant (e.g. peptides) to donate an electron which can reduce Fe3+⁄ferric cyanide complex to the ferrous form [39]. The reducing ability of a compound may serve as a significant indicator of its potential antioxidant activity [68,69]. Several reports have revealed that there is a direct correlation between antioxidant activities andreducing power of certain bioactive compounds. In particular, assays that establish the ability of various bean protein hydrolysates and their membrane fractions to serve as reducing agents or antioxidants are available in literature [3,26,67]. During the assay for reducing power, an oxidant probe accepts electron from the antioxidant analyte (e.g. peptides) and becomes converted into the reduced probe which is colored [70]. When measured at 700 nm, an increase in absorbance indicates better reducing power of the test sample [70]. As shown in Figure 4B, the KBH and its peptide fractions showed different levels of potency in reducing Fe3+ to the ferrous form when compared to the GSH standard. The reducing power of the <1 kDa fraction was the highest, followed by 5–10 kDa, KBH, then 3–5 and 1–3 kDa, respectively (Figure 4B). The reducing power of alcalase–hydrolyzed KBH and its peptide fractions was increased with increasing peptide concentrations. A concentration–dependency of reducing power has also been observed for chickpea protein hydrolysate [18], Rumex crispus L. extracts [39] and fermented soy protein hydrolysate [71]. The highest activity of the <1 kDa fraction may be due to higher content of the hydrophobic amino acids as evident in Table 1. Ajibola et al. [38] also observed that the 1 kDa fraction from African yam bean seed protein hydrolysate had significantly higher (p<0.05) reducingpower when compared to those of unfractionated hydrolysate and the 1-3, 3-5, 5-10 kDa peptide fractions. Previous works have suggested that smaller size peptides exhibited better reducing power than high molecular weight fractions [38,59]. However, as reported by Wu etal. [72] and Pownall et al. [41] for protein hydrolysates of mackerel and pea seed protein hydrolysate fractions, respectively, the present results relate more to the total hydrophobic amino acid contents of the fractions than the peptide size. Li et al. [18] reported similar observation (effects of peptide hydrophobicity) for chickpea proteinhydrolysate fractions.
Inhibition of linoleic acid peroxidation
Lipid peroxidation (LPO) is of serious concern to the food industry because it results in subsequent development of undesirable offflavours, odours, dark colours and potentially toxic reaction [73]. More importantly to human health is that the free radicals that result from LPO can cause damage to cellular macromolecules through oxidative modifications of the genome, proteins, structural carbohydrates, and lipids, thereby affecting normal physiological functions [74]. Antioxidants are capable of inhibiting oxidation of lipids. The demand for natural antioxidants, such as those derived from food, has recently increased because of questions about the long–term safety and negative consumer perception of synthetic antioxidants. The antioxidative activity of various fractions of KBH on the oxidation of linoleic acid is shown in Figure 5. In all the samples, especially in the control, the absorbance increased to a maximum and then decreased gradually. Chen et al. [75] and Chen et al. [76] made similar observations for the antioxidant activity of designed peptides and peanut protein hydrolysate, respectively. The decrease in absorbance after the 4th day may be because hydroperoxides are usually unstable and they will gradually decompose into secondary metabolites as the experiment rogressed. The products formed during the oxidation reacts with the iron (II) sulphate to form iron (III) sulphate, which in turn will react with ammonium thiocyanate to form a colored complex of ferric thiocyanate; therefore, absorption intensity is directly related to degree of linoleic acid oxidation [41,75]. The current data suggest that mostof the linoleic acid molecules become oxidized after about four days, after which peroxide formation is reduced and hence the decreased absorption as the experiment progressed. When compared with the control, addition of peptides exhibited a noticeable antioxidant property to inhibit the LPO as shown by the reduction in absorption intensity of the incubated sample solutions to various degrees (Figure 5). Generally, the rate of chain reaction initiated during LPO is inhibited by the ability of antioxidants to reduce and transform the reactive end products to a more stable form [77]. Hydrophobic amino acid residues such as Tyr, Met, His, Lys, and Trp have been shown to have strong antioxidant activity against lipid derived–radicals due to the ability of hydrophobic amino acids to interact with the lipids [41,75]. In the current study, the abilities of the fractions to inhibit the LPO corresponded with the amount of hydrophobic amino acid residues in the peptide fractions. The hydrolysates with higher hydrophobic amino acid residues possessed stronger antioxidant activity because hydrophobicity of the compounds was important for interaction with and protection of the hydrophobic linoleic acid from peroxidation. Consequently, the <1 and 5–10 kDa fractions exhibited the highest inhibitory activity, followed by 3–5 kDa and then 1–3 kDa. Although, the <1 kDa fraction had the highest hydrophobic amino acid residues, histidine containing peptides which was highest in the 5–10 kDa fraction may have also played key role in the inhibition of linoleic acid oxidation. This is because of histidine’s ability to trap lipid peroxyradical [78]. GSH showed the highest inhibitory activity in the first five days, after which inhibitory activity was similar to the 1–3 and 5–10 kDa peptides. Pownall et al. [41] reported similar trend in GSH inhibitory activity when compared with pea protein hydrolysate and its membrane fractions. Samples (1 mg⁄ml) of pea protein hydrolysate and its high performance liquid chromatography fractions as reported by Pownall et al. [41], exhibited stronger ability to inhibit linoleic acid oxidation over 7 days than observed for the KBH fractions in the current study. However, peanut protein hydrolysate even at 2 mg⁄ml, exhibited negligible inhibitory activity of linoleic acid peroxidation [76] when compared to the observations in our study.

Figure 5: Plate 2: Purple Star Apple
Conclusion
The <1 and 5–10 kDa fractions exhibited significantly highest (p<0.05) renin inhibition and the ability to scavenge DPPH free radical, inhibit peroxidation of linoleic acid and reduce Fe3+ to Fe2+. Therefore, the fractions with <1 and 5–10 kDa peptides showed better potential as antihypertensive and antioxidant peptides, probably due to their slightly higher contents of hydrophobic amino acids. However, it is also well known that smaller size peptides may be more bioactive because of the higher probability for increased rate of intestinal absorption (without structural degradation) and entry into cells when compared with the bigger size peptides. In addition to the small size, the high content of hydrophobic amino acids may also contribute to increase absorption rate for the <1 kDa peptides and make them better bioactive agents than the longer 5–10 kDa peptides.
Acknowledgements
This work was funded through a Discovery grant from the Natural Sciences and Engineering Research Council of Canada (NSERC) to sDr R.E. Aluko.
References
- Hartmann R, Meisel H . Food-derived peptides with biological activity: from research to food applications. Curr Opin Biotechnol. 2007; 18: 163-169.
- Möller NP, Scholz-Ahrens KE, Roos N, Schrezenmeir J . Bioactive peptides and proteins from foods: indication for health effects. Eur J Nutr. 2008; 47: 171-182.
- Xiong YL (2010) Antioxidant peptides. In: Bioactive proteins and peptides as functional foods and nutraceuticals (Mine Y, Li-Chan ECY, Jiang B, eds), pp29-39. New York; Wiley-Blackwell.
- Ngo DH, Vo TS, Ngo DN, Wijesekara I, Kim SK . Biological activities and potential health benefits of bioactive peptides derived from marine organisms. Int J Biol Macromol. 2012; 51: 378-383.
- Udenigwe CC, Aluko RE . Food protein-derived bioactive peptides: production, processing, and potential health benefits. J Food Sci. 2012; 77: R11-24.
- Li H, Aluko RE . Identification and Inhibitory Properties of Multifunctional Peptides from Pea Protein Hydrolysate. J Agric Food Chem. 2010;
- Mäkinen S, Johannson T, Vegarud Gerd E, Pihlava JM, Pihlanto A. Angiotensin I-converting enzyme inhibitory and antioxidant properties of rapeseed hydrolysates. J Funct Foods 2012; 4: 575-583.
- Udenigwe CC, Lu Y-L, Han C-H, Hou W-C, Aluko RE. Flaxseed protein-derived peptide fractions: Antioxidant properties and inhibition of lipopolysaccharide-induced nitric oxide production in murine macrophages. Food Chem. 2009; 116: 277-284.
- Udenigwe CC, Adebiyi AP, Doyen A, Li H, Bazinet L, et al. Low molecular weight flaxseed protein-derived arginine-containing peptides reduced blood pressure of spontaneously hypertensive rats faster than amino acid form of arginine and native flaxseed protein. Food Chem. 2012; 132: 468-475.
- Pe&nTilde;ta-Ramos EA, Xiong YL. Antioxidant activity of soy protein hydrolysates in a liposomal system. J Food Sci. 2002; 67: 2952-2956.
- Matsui T, Tanaka M (2010) Antihypertensive peptides and their underlying mechanisms. In: Bioactive proteins and peptides as functional foods and nutraceuticals (Mine Y, Li-Chan ECY, Jiang B, ed), pp43-53. New York: Wiley-Blackwell.
- Nakahara T, Sano A, Yamaguchi H, Sugimoto K, Chikata H . Antihypertensive effect of peptide-enriched soy sauce-like seasoning and identification of its angiotensin I-converting enzyme inhibitory substances. J Agric Food Chem. 2010; 58: 821-827.
- Hooper NM . Angiotensin converting enzyme: implications from molecular biology for its physiological functions. Int J Biochem. 1991; 23: 641-647.
- Tenenbaum A, Grossman E, Shemesh J, Fisman EZ, Nosrati I . Intermediate but not low doses of aspirin can suppress angiotensin-converting enzyme inhibitor-induced cough. Am J Hypertens. 2000; 13: 776-782.
- Abassi Z, Winaver J, Feuerstein GZ. The biochemical pharmacology of renin inhibitors: implications for translational medicine in hypertension, diabetic nephropathy and heart failure: expectations and reality. Biochem Pharmacol. 2009; 78: 933-940.
- Kitts DD, Weiler K . Bioactive proteins and peptides from food sources. Applications of bioprocesses used in isolation and recovery. Curr Pharm Des. 2003; 9: 1309-1323.
- Matsui T, Matsumoto K (2006) Antihypertensive peptides from natural resources. In: Lead molecules from natural products Discovery and new trends (Khan THM, Ather A, ed), pp255-271. Amsterdam: Elsevier.
- Li Y, Jiang B, Zhang T, Mu W, Liu J. Antioxidant and free radical-scavenging activities of chickpea protein hydrolysate (CPH). Food Chem. 2008; 106: 444-450.
- Nazeer RA, Srividhya TS. Antioxidant peptides from the protein hydrolysates of Conus betulinus. Int J Peptide Res Therap. 2011; 17: 231-237.
- Aluko RE, Monu E. Functional and bioactive properties of quinoa seed protein hydrolysates. J Food Sci. 2003; 68: 1254-1258.
- Amarowicz R, Shahidi F. Antioxidant activity of peptide fractions of capelin protein hydrolysates. Food Chem. 1997; 58: 355-359.
- Cumby N, Zhong Y, Naczk M, Shahidi F. Antioxidant activity and water-holding capacity of canola protein hydrolysates. Food Chem. 2008; 109: 144-148.
- You S-J, Udenigwe CC, Aluko RE, Wu J. Multifunctional peptides from egg white lysozyme. Food Res Int. 2010; 43: 848-855.
- Megías C, Pedroche J, Yust MM, Julio Girón-Calle, Millán F, et al. Affinity purification of copper chelating peptides from chickpea protein hydrolysates. J Agric Food Chem. 2007; 55: 3949-3954.
- Yust Mdel M, Millán-Linares Mdel C, Alcaide-Hidalgo JM, Millán F, Pedroche J . Hypocholesterolaemic and antioxidant activities of chickpea (Cicer arietinum L.) protein hydrolysates. J Sci Food Agric. 2012; 92: 1994-2001.
- Zhang T, Li Y, Miao M, Jiang B. Purification and characterisation of a new antioxidant peptide from chickpea (Cicer arietium L.) protein hydrolysates. Food Chem. 2011; 128: 28-33.
- Segura Campos MR, Chel Guerrero LA, Betancur Ancona DA . Angiotensin-I converting enzyme inhibitory and antioxidant activities of peptide fractions extracted by ultrafiltration of cowpea Vigna unguiculata hydrolysates. J Sci Food Agric. 2010; 90: 2512-2518.
- Darmawan R, Bringe NA, de Mejia EG . Antioxidant capacity of alcalase hydrolysates and protein profiles of two conventional and seven low glycinin soybean cultivars. Plant Foods Hum Nutr. 2010; 65: 233-240.
- Yin S, Tang C, Yang X, Wen Q, Qi J. Surface charge and conformational properties of phaseolin, the major globulin in red kidney bean (Phaseolus vulgaris L): Effect of pH. Int J Food Sci Technol. 2011; 46: 1628-1635.
- Mundi S, Aluko RE. Physicochemical and functional properties of kidney bean albumin and globulin protein fractions. Food Res Int. 2012; 48: 299-306.
- Markwell MA, Haas SM, Bieber LL, Tolbert NE . A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal Biochem. 1978; 87: 206-210.
- Bidlingmeyer BA, Cohen SA, Tarvin TL . Rapid analysis of amino acids using pre-column derivatization. J Chromatogr. 1984; 336: 93-104.
- Gehrke CW, Wall LL, Absheer JS. Sample preparation for chromatography of amino acids: Acid hydrolysis of proteins. J Assoc Official Anal Chem. 1985; 68: 811-821.
- Landry J, Delhaye S. Simplified procedure for the determination of tryptophan of foods and feedstuffs from barytic hydrolysis. J Agric Food Chem. 1992; 40: 776-779.
- Hayakawa S, Nakai S. Relationships of hydrophobicity and net charge to the solubility of milk and soy proteins. J Food Sci. 1985; 50: 486-491.
- Udenigwe CC, Lin Y, Hou W, Aluko RE. Kinetics of the inhibition of renin and angiotensin I-converting enzyme by flaxseed protein hydrolysate fractions. J Funct Foods 2009; 1: 199-207.
- Hou WC, Lee MH, Chen HJ, Liang WL, Han CH . Antioxidant activities of dioscorin, the storage protein of yam (Dioscorea batatas Decne) tuber. J Agric Food Chem. 2001; 49: 4956-4960.
- Ajibola CF, Fashakin JB, Fagbemi TN, Aluko RE . Effect of Peptide Size on Antioxidant Properties of African Yam Bean Seed (Sphenostylis stenocarpa) Protein Hydrolysate Fractions. Int J Mol Sci. 2011; 12: 6685-6702.
- Yildirim A, Mavi A, Kara AA . Determination of antioxidant and antimicrobial activities of Rumex crispus L. extracts. J Agric Food Chem. 2001; 49: 4083-4089.
- [No authors listed] . Energy and protein requirements. Report of a joint FAO/WHO/UNU Expert Consultation. World Health Organ Tech Rep Ser. 1985; 724: 1-206.
- Pownall TL, Udenigwe CC, Aluko RE . Amino acid composition and antioxidant properties of pea seed ( Pisum sativum L.) enzymatic protein hydrolysate fractions. J Agric Food Chem. 2010; 58: 4712-4718.
- Wu W, Williams WP, Kunkel ME, Acton JC, Huang Y, et al. Amino acid availability and availability-corrected amino acid score of red kidney beans (Phaseolus vulgaris L.). J Agric Food Chem. 1996; 44: 1296-1301.
- Wu J, Aluko RE, Muir AD . Production of angiotensin I-converting enzyme inhibitory peptides from defatted canola meal. Bioresour Technol. 2009; 100: 5283-5287.
- Pripp AH, Isaksson T, Stepaniak L, Sørhaug T. Quantitative structure-activity relationship modelling of ACE-inhibitory peptides derived from milk proteins. Eur Food Res Technol. 2004; 219: 579-583.
- u J, Aluko RE, Nakai S . Structural requirements of Angiotensin I-converting enzyme inhibitory peptides: quantitative structure-activity relationship study of di- and tripeptides. J Agric Food Chem. 2006; 54: 732-738.
- Voronov SV, Skirgello OE, Troshina NN, Orlova MA, Kost OA . A hydrophobic site on the surface of the angiotensin-converting enzyme molecule. Biochemistry (Mosc). 2002; 67: 553-557.
- Wu WU, Hettiarachchy NS, Qi M. Hydrophobicity, solubility, and emulsifying properties of soy protein peptides prepared by papain modification and ultrafiltration. J Am Oil Chem Soc. 1998; 75: 845-850.
- Molina Ortiz SE, Wagner JR. Hydrolysates of native and modified soy protein isolates: Structural characteristics, solubility and foaming properties. Food Res Int. 2002; 35: 511-518.
- Wang J-S, Zhao M, Zhao Q-Z, Jiang Y-M. Antioxidant properties of papain hydrolysates of wheat gluten in different oxidation systems. Food Chem. 2007; 101: 1658-1663.
- Jang JH, Jeong SC, Kim JH, Lee YH, Ju YC . Characterisation of a new antihypertensive angiotensin I-converting enzyme inhibitory peptide from Pleurotus cornucopiae. Food Chem. 2011; 127: 412-418.
- Udenigwe CC, Li H, Aluko RE . Quantitative structure-activity relationship modeling of renin-inhibiting dipeptides. Amino Acids. 2012; 42: 1379-1386.
- Yuan L, Wu J, Aluko RE, Ye X . Kinetics of renin inhibition by sodium houttuyfonate analogs. Biosci Biotechnol Biochem. 2006; 70: 2275-2280.
- Li GH, Le GW, Liu H, Shi YH. Mung-bean protein hydrolysates obtained with alcalase exhibit angiotensin I-converting enzyme inhibitory activity. Food Sci Tech Int. 2005; 11: 281-287.
- Valdez-Ortiz A, Fuentes-Gutiérrez CI, Germán-Báez LJ, Gutiérrez-Dorado R, Medina-Godoy S. Protein hydrolysates obtained from azufrado (sulphur yellow) beans (Phaseolus vulgaris): Nutritional, ACE-inhibitory and antioxidative characterization. LWT - Food Sci Technol. 2012; 46: 91-96.
- Ruiz-Ruiz J, Dávila-Ortíz G, Chel-Guerrero L, Betancur-Ancona D. Angiotensin I-converting enzyme inhibitory and antioxidant peptide fractions from hard-to-cook bean enzymatic hydrolysates. J Food Biochem. 2013; 37: 26-35.
- Corrêa AP, Daroit DJ, Coelho J, Meira SM, Lopes FC . Antioxidant, antihypertensive and antimicrobial properties of ovine milk caseinate hydrolyzed with a microbial protease. J Sci Food Agric. 2011; 91: 2247-2254.
- Girgih AT, Udenigwe CC, Aluko RE. In vitro antioxidant properties of hemp seed (Cannabis sativa L.) protein hydrolysate fractions. J Am Oil Chem Assoc. 2011; 88: 381-389.
- Bamdad F, Wu J, Chen L. Effects of enzymatic hydrolysis on molecular structure and antioxidant activity of barley hordein. J Cereal Sci. 2011; 54: 20-28.
- Bougatef A, Hajji M, Balti R, Lassoued I, Triki-Ellouz Y, et al. Antioxidant and free radical-scavenging activities of smooth hound (Mustelus mustelus) muscle protein hydrolysates obtained by gastrointestinal proteases. Food Chem. 2009; 114: 1198-1205.
- Du J, Gebicki JM . Proteins are major initial cell targets of hydroxyl free radicals. Int J Biochem Cell Biol. 2004; 36: 2334-2343.
- Yen G, Hsieh P. Antioxidative activity and scavenging effects on active oxygen of xylose-lysine maillard reaction products. J Sci Food Agric. 1995; 67: 415-420.
- Morita A, Kimura M, Itokawa Y. The effect of aging on the mineral status of female mice. Biol Trace Element Res. 1994; 42: 165-177.
- Takahashi S, Takahashi I, Sato H, Kubota Y, Yoshida S . Age-related changes in the concentrations of major and trace elements in the brain of rats and mice. Biol Trace Elem Res. 2001; 80: 145-158.
- Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR . Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004; 5: 863-873.
- Arosio P, Levi S . Ferritin, iron homeostasis, and oxidative damage. Free Radic Biol Med. 2002; 33: 457-463.
- Nam KA, You SG, Kim SM . Molecular and physical characteristics of squid (Todarodes pacificus) skin collagens and biological properties of their enzymatic hydrolysates. J Food Sci. 2008; 73: C249-255.
- Carrasco-Castilla J, Hernández-Álvarez AJ, Jiménez-Martínez C, Jacinto-Hernández C, Alaiz M, et al. Antioxidant and metal chelating activities of Phaseolus vulgaris L. var. jamapa protein isolates, phaseolin and lectin hydrolysates. Food Chem. 2012; 131: 1157-1164.
- Meir S, Kanner J, Akiri B, Philosoph-Hadas S. Determination and involvement of aqueous reducing compounds in oxidative defense systems of various senescing leaves. J Agric Food Chem. 1995; 43: 1813-1819.
- Oktay M, Gülçin I, Küfreviolu ÖI. Determination of in vitro antioxidant activity of fennel (Foeniculum vulgare) seed extracts. LWT - Food Sci Technol. 2003; 36: 263-271.
- Berker KI, Güçlü K, Tor I, Apak R. Comparative evaluation of Fe(III) reducing power-based antioxidant capacity assays in the presence of phenanthroline, batho-phenanthroline, tripyridyltriazine (FRAP), and ferricyanide reagents. Talanta 2007; 72: 1157-1165.
- Amadou I, Le G-W, Shi Y-H, Jin S. Reducing, radical scavenging, and chelation properties of fermented soy protein meal hydrolysate by lactobacillus plantarum LP6. Int J Food Prop. 2011; 14: 654-665.
- Wu H, Chen H, Shiau C. Free amino acids and peptides as related to antioxidant properties in protein hydrolysates of mackerel (Scomber austriasicus). Food Res Int. 2003; 36: 949-957.
- Sakanaka S, Tachibana Y, Ishihara N, Juneja LR. Antioxidant activity of egg-yolk protein hydrolysates in a linoleic acid oxidation system. Food Chem. 2004; 86: 99-103.
- Romero FJ, Bosch-Morell F, Romero MJ, Jare&nTilde;o EJ, Romero B . Lipid peroxidation products and antioxidants in human disease. Environ Health Perspect. 1998; 106 Suppl 5: 1229-1234.
- Chen H-M, Muramoto K, Yamauchi F, Nokihara K. Antioxidant activity of designed peptides based on the antioxidative peptide isolated from digests of a soybean protein. J Agric Food Chem. 1996; 44: 2619-2623.
- Chen G-T, Zhao L, Zhao L-Y, Cong T, Bao S. In vitro study on antioxidant activities of peanut protein hydrolysate. J Sci Food Agric. 2007; 87: 357-362.
- Sowndhararajan K, Siddhuraju P, Manian S. Antioxidant activity of the differentially processed seeds of jack bean (Canavalia ensiformis L. DC). Food Sci Biotechnol. 2011; 20: 585-591.
- Erdmann K, Cheung BW, Schröder H . The possible roles of food-derived bioactive peptides in reducing the risk of cardiovascular disease. J Nutr Biochem. 2008; 19: 643-654.