
Case Series
Austin J Radiol. 2017; 4(2): 1067.
The Role of Imaging in Mayer-Rokitansky-Küster-Hauser Syndrome: A Report of Two Cases and Literature Review
Farate A¹*, Ahmadu MS¹, Tela UM², Shettima ZB¹ and Bukar M³
¹Department of Radiology, University of Maiduguri Teaching Hospital, Nigeria
²Division of Urology, Department of Surgery, University of Maiduguri Teaching Hospital, Nigeria
³Department of Obstetrics and Gynaecology, University of Maiduguri Teaching Hospital, Nigeria
*Corresponding author: Farate A, Department of Radiology, University of Maiduguri Teaching Hospital, P. M. B. 1414 Maiduguri, Borno State, Nigeria
Received: April 05, 2017; Accepted: May 15, 2017; Published: May 26, 2017
Abstract
Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome is a rare condition that results from the disturbance of embryonic paramesonephric (Mullerian) duct development giving rise to varying degrees of malformation of reproductive organs and in some cases structures outside of reproductive system such as renal and skeletal abnormalities. The authors describe two female patients with normal (46, XX) karyotype, normal external genitalia and well-developed secondary sexual characteristics who presented with primary amenorrhoea.
Keywords: Mullerian defects; Mayer-Rokitansky-Kuster-Hauser syndrome; Primary amenorrhoea; Imaging
Introduction
Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome results from developmental failure of part or whole of Müllerian ducts that is characterised by partial or complete absence of the uterus with absent or shallow vaginal pouch. Patients with MRKH syndrome have normal karyotype (46, XX), external genitalia and secondary sexual characteristics (pubic hair and breast development) [1-3].
The MRKH syndrome is broadly classified into two types. Type I (typical form) is characterised by abnormalities that are confined to the reproductive system and type II or MURCS (Müllerian duct aplasia, renal dysplasia, and cervicothoracic somite anomalies) association (atypical form), if there is an additional abnormality, most commonly renal (unilateral agenesis, ectopic kidneys, horseshoe kidneys) and skeletal (Klippel-Feil anomaly, fused vertebra—particularly cervical vertebra and scoliosis) [1-3].
We report on the two distinct forms of MRKH females who presented with primary amenorrhoea and normal secondary sexual characteristics. The first patient, an 18-year-old, had an isolated abnormality of the reproductive system (type I MRKH syndrome) and the second patient, a 25-year-old, in addition to reproductive system abnormality had associated horseshoe kidney, cervical vertebral fusion (Klippel-Feil anomaly) and lumbar spine scoliosis (type II MRKH syndrome or MURCS association) on further imaging evaluation. This report highlights the role and benefit of imaging in the differential diagnosis and classification of patients presenting with this disorder.
Case Presentation
Case 1
An 18-year-old girl referred for ultrasound examination on account of primary amenorrhoea. She had no other medical problem. Her 14-year-old immediate younger sister had attained menarche and experienced normal and regular menstrual cycle. No family history of similar problem. Patient’s mother did not use any medications that are known to be teratogenic during pregnancy. The patient had begun pubertal development at 12-years-old and thelarche at 10-years-old.
At physical examination, the height measured 160 cm and weight measured 45kg. The secondary sexual characteristics such as breast development (Tanner stage-5), axillary and pubic hair was compatible with her chronological age. External genitalia, urethra, and vaginal orifice were normal. Digital vaginal and speculum examinations were deferred because of virginity.
At transabdominal ultrasound, both ovaries were seen with no identifiable uterine tissue (Figure 1a & b). Magnetic Resonance Imaging (MRI) revealed normal ovaries containing multiple subcentimeter follicles (Figure 1c & d). The ovaries measured 20 mm x 24 mm on the right and 23 mm x 27 mm on the left. The uterus was absent (Figure 2). Both kidneys were in their normal location and Intravenous Pyelogram (IVP) did not show any pelvicalyceal, ureteral or urinary bladder abnormality (Figure 3). Radiographic examination of the spine was normal. Total blood count and relevant serum biochemistry values were within normal limits.
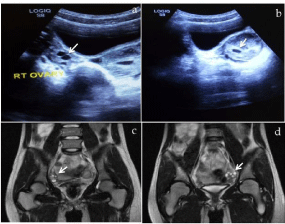
Figure 1: Pelvic sonogram (top) and coronal T2-weighted MR images
(bottom). The top images demonstrates the normally located ovaries, the
right ovary (a) and the left ovary (b) with corresponding MRI images at the
bottom showing the right ovary (c) and the left ovary (d) both containing
subcentimeter follicles—arrows.
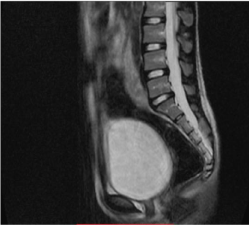
Figure 2: Sagittal T1-weighted MR.There is clearly absence of uterus or
rudimentary uterine horn.
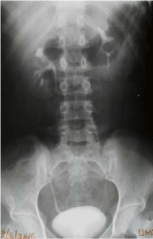
Figure 3: An intravenous urography. The pelvicalyces, ureters and the
urinary bladder are normal with no associated urinary tract abnormality.
Case 2
A 25-year-old married female patient presented with primary amenorrhoea and dyspareunia. She had no other medical ailments. There was no family history of similar condition. At physical examination, the height measured 153 cm and weight measured 46 kg. The secondary sexual characteristics such as breast development (Tanner stage-5), axillary and pubic hair was normal. External genitalia, urethra, and vaginal orifice were also normal. Digital vaginal examination revealed a small vagina, barely measured 3 cm in length, with a blind end.
At transabdominal US, the uterus and ovaries were not visualised. Both renal fossae were empty; however, a low-lying horseshoe kidney was demonstrated (Figure 4a). MRI confirmed the presence of a horseshoe kidney at the level of L4 to S1 vertebrae (Figure 4b & c) with no identifiable uterine tissue (Figure 5a). Both ovaries were visualised containing subcentimeter follicles; they measured 18 mm x 28 mm on the right and 17 mm x 20 mm on the left. The right ovary was displaced superolaterally to the pelvic sidewall; the left ovary appeared to be in normal location (Figure 5b & c). At IVP, ureters were correspondingly short and inserted normally into the urinary bladder (Figure 6a). Radiograph of the cervical spine showed fusion of C5 to C7 vertebrae (Figure 6b). Gentle scoliosis and rotation of lumbar spine were also evident (Figure 6c). Complete blood count and relevant serum biochemistry values were normal.
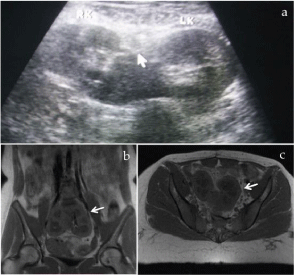
Figure 4: Ultrasound, coronal T1-weighted and axial T1-weighted MR
images. Transabdominal sonogram (a) demonstrates a “horse-shoe” kidney
with corresponding MR images in coronal (b) and axial (c) planes clearly
demonstrating a low lying “horse-shoe” kidney.
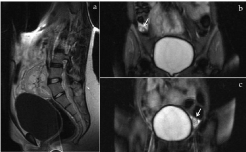
Figure 5: Sagittal T1-weighted and coronal T2-weighted MR images.
The uterus or its rudimentary uterine horn is absent (a). The right ovary
is superolaterally displaced (b) and the left ovary appears to be in normal
anatomic location (c).
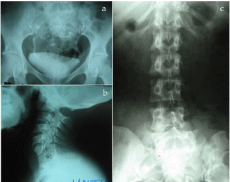
Figure 6: Contrast and plain radiographs. There is an abnormally located
pelvic kidney(s) with correspondingly short ureters (a). Associated fusion of
the lower cervical vertebra— Klippel-Feil anomaly (b) and scoliosis of the
lumbar spine (c) are evident.
Discussion
Mayer-Rokitansky-Küster-Hauser syndrome is the second most common cause of primary amenorrhoea after gonadal dysgenesis [4]. It has an incidence of 1: 4000 to 5000 female births [3]. The aetiology of this syndrome is unknown but genetic predisposition with an autosomal dominant mode of inheritance has been identified [1,2]. Karl Mayer (1829) and Von Rokitansky (1838) described the syndrome as uterovaginal agenesis due to abnormal development of uterine ducts [3,5,6]. Hermann Küster (1910) identified the urological associations and G. A. Hauser (1961) distinguished MRKH females from testicular feminisation [3,5].
The clinical diagnosis of MRKH syndrome is often delayed until puberty when a patient presents with primary amenorrhoea. Upon physical examination, the female secondary sexual characteristics are normal due to normal ovarian function with no sign or biochemical evidence of androgen excess [1,2]. While the diagnosis of MRKH syndrome can be established on clinical ground, further evaluation using imaging, laparoscopy and karyotyping is necessary to confirm the diagnosis and to detect the associated anomalies.
The differential diagnosis that can be mistaken for MRKH syndrome is testicular feminisation syndrome (androgen insensitivity syndrome). Patients with testicular feminisation syndrome have 46, XX karyotype in which the affected males have female external genitalia, female breast development, short blind-ending vagina, absent uterus and female adnexa, abdominal or inguinal testes and no axillary or pubic hair [1-3]. Other differential diagnoses are gonadal dysgenesis, agenesis of the vagina and uterus, imperforate hymen and transverse lower vaginal septum, but the uterus and tubes are normal in these conditions.
Ultrasonography is the first line imaging modality of choice in the diagnosis of MRKH syndrome because it is simple to perform, readily available, non-invasive and does not make use of ioinsing radiation. Ultrasound scan may demonstrate the absence of the uterine tissue between the urinary bladder and rectum. The lack of utero-ovarian ligaments or incomplete ovarian descent in patients with MRKH syndrome may result in high pelvic location of the ovaries making sonographic detection difficult [5-7]. In one of our cases, the right ovary was observed above the normal location. Ultrasound is also useful for the detection of associated renal anomaly.
MRI should be performed when ultrasound findings are inconclusive, since failure to clearly identify the uterus or Müllerian rudiments or ovaries does not suggest their absence [1,2]. The multiplanar imaging and high soft tissue resolution of MRI makes it suitable modality to clearly assess the pelvic anatomy and possible pathological conditions. It is imperative to detect rudimentary uterus between bladder and rectum in sagittal T2-weighted sequences. MRI is also important for detecting high pelvic location of the ovaries and to reveal associated urinary tract anomalies. Laparoscopy is indicated only in cases where the evaluation by ultrasound and MRI is inconclusive and provided this method allows the definition of a therapeutic strategy.
Plain radiographs are performed for associated skeletal abnormalities and contrast studies such as IVP to assess the renal tract anomalies. Although computed tomography provides information about congenital anomalies, it is not routinely performed because of ionising radiation.
The management of MRKH syndrome requires a multidisciplinary team approach including gynaecologists, radiologists, clinical psychologists and other specialists where applicable. Extensive counselling and emotional support is critical at the time of diagnosis and later in life as these patients may have intense psychological problem with even tendency to commit suicide [8]. The definitive treatment consists of the creation of a neovagina, via surgical and nonsurgical methods. Non-surgical technologies are based on the formation of a neovagina by vaginal dilators (Frank’s Technology). The most commonly used surgical techniques are: sigmoid neovaginoplasty, which uses as a neovagina a segment of sigmoid colon and the Vecchietti operation, in which a neovagina is created by gradual stretching of the vaginal skin [1-3]. The two techniques mentioned above can be performed laparoscopically there by reducing the associated morbidity and hospital stay compared to open surgery.
The ability to reproduce is one of the most emotionally devastating aspects of MRKH syndrome. Because the ovaries function normally, in vitro fertilisation and surrogacy are possible means of producing genetically related offspring using the woman’s own eggs. This can be achieved via transabdominal or rarely, laparoscopic egg retrieval. An attempt at uterine transplantation in human was unsuccessful despite several promising animal studies. Research is focusing on such novel therapies and the genetic aetiology and pattern of inheritance of MRKH syndrome [3].
Urinary tract abnormalities associated with MRKH syndrome usually do not require any urologic surgical intervention. Rarely, patients may have urethral coitus as a result of vaginal agenesis associated with this syndrome [9]. This leads to urethral dilatation, incontinence and recurrent urinary tract infections and urethroplasty becomes mandatory [10].
Conclusion
Radiologic imaging is crucial in the diagnosis, classification and management of MRKH syndrome. MRI allows an accurate evaluation of the uterine aplasia, as well as clear visualisation of the rudimentary horns and ovaries owing to excellent soft tissue resolution. Identification of normal and functional ovaries is key to distinguishing MRKH females from testicular feminisation. Plain radiography assesses the associated skeletal involvement with ultrasound and contrast IVP to determine the associated urologic alterations.
Competing Interest
The authors declare that they have no financial or personal relationship(s) that may have inappropriately influenced them in writing this manuscript.
References
- Morcel K, Camborieux L. Programme de Recherchessur les Aplasies Mülleriennes (PRAM), Guerrier D. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Orphanet J of Rare Dis. 2007; 2: 13.
- Pizzo A, Lagana AS, Sturlese E, Retto G, Retto A, De Dominici, et al. Mayer- Rokitansky-Kuster-Hauser syndrome: embryology, genetics and clinical and surgical treatment. Obstet Gynecol. 2013; 2013: 628-717.
- Valappil S, Chetan U, Wood N, Garden A. Mayer–Rokitansky–Kuster–Hauser syndrome: diagnosis and management. The Obstetrician & Gynaecologist. 2012; 14: 93–98.
- Kwon SK, Chae HD, Lee KH, Kim SH, Kim CH, Kang BM. Causes of amenorrhoea in Korea: Experience of a single large centre. Clin Exp Reprod Med. 2014; 41: 29-32.
- Kara T, Acu B, Beyhan M, Gokce E. MRI in the diagnosis of Mayer- Rokitansky-Kuster-Hauser syndrome. DiagnInterv Radiol. 2013; 19: 227-232.
- Bozkurt DK, Bozkurt M, Sahin L. Magnetic Resonance Imaging and Clinical Features in Mayer-Rokitansky-Kuster-Hauser Syndrome. J Med Cases. 2014; 5: 182-185.
- Lalitha Kumari G, Panil Kumar BE, Ramanappa MV, Sreedhar Reddy B, Reddy MM. Role of MRI in Evaluation of MRKH Syndrome. J of Evidence Based Med & Healthcare. 2015; 2: 8555-8560.
- Gupta M, Kharb V. MRKH Syndrome: Psychological disturbances and suicide. J Indian Acad Forensic Med. 2012; 34: 86-88.
- Rouzi AA. Urethral sex in a woman with previously undiagnosed Mayer- Rokitansky-Kuster-Hauser syndrome. Clin Exp Obstet Gynecol. 2013; 40: 452-453.
- Sharifiaghdas F, Daneshpajooh A, Sohbati S, Mirzaei M. An unusual cause of urinary incontinence: Urethral coitus in a case of Mayer-Rokitansky-Kuster- Hauser syndrome. Investig Clin Urol. 2016; 57: 367-371.