
Special Article - Radiology Case Reports
Austin J Radiol. 2019; 6(3): 1097.
Pulmonary Langerhans Cell Histiocytosis: Trapped Case
Ozkavruk Eliyatkin N1*, Tataroglu C1, Karaman CZ2 and Kanlioglu Kuman N3
¹Department of Pathology, Adnan Menderes University, Turkey
²Department of Radiology, Adnan Menderes University, Turkey
³Department of Chest Surgery, Adnan Menderes University, Turkey
*Corresponding author: Nuket Ozkavruk Eliyatkin, Department of Pathology, Faculty of Medicine, Adnan Menderes University, 09100 Aydin, Turkey
Received: July 08, 2019;Accepted: July 23, 2019;Published: July 30, 2019
Abstract
Langerhans Cell Histiocytosis (LCH) is a rarely seen disease and has a very large spectrum varying between solitary bone lesions and involvement of many organs or systems. Pulmonary involvement in LCH may be a component of a multisystem disease or it may affect only lungs. Herein, we are presenting a 35-year-old male patient with LCH confined to lungs which progressed with the development of spontaneous pneumothorax that might be overlooked during histopathological evaluation because of its development in the interstitial space without nodular formation.
Keywords: Langerhans cell histiocytosis; pulmonary
Introduction
Langerhans Cell Histiocytosis (LCH) is a rarely seen disease characterized by nodules consisting of cells having the phenotype of Langerhans cells, and various inflammatory cells. It has a very large spectrum varying between solitary bone lesions and involvement of many organs or systems [1].
Pulmonary involvement in LCH may be a component of a multisystem disease or it may affect only lungs. Pulmonary Langerhans Cell Histiocytosis (PLCH) is generally confined to lungs. Basically PLCH is a form of interstitial pulmonary disease, and it is accepted as a disease different from systemic LCH. The symptoms of this disease are very variable and may include dyspnea, chest pain, hemoptysis, and spontaneous pneumothorax [2].
Herein, we are presenting a 35-year-old male patient with LCH confined to lungs which progressed with the development of spontaneous pneumothorax that might be overlooked during histopathological evaluation because of its development in the interstitial space without nodular formation.
Case Report
A 35-year-old male. Smoking stories are available for 10 years. He has quit smoking for the last 5 years. On 05.12.2016, the patient came to the emergency service. There was an increased chest pain in battle style with movement. Except for a reduction in respiratory sounds in the left lung, physical examination was unusual in the patient with COPD. Imaging findings with pneumothorax pre-diagnosis revealed that the lower lobes of both lungs were preserved (Figure 1). Due to the developing pneumothorax, wedge resection of the lower left superior segment was performed.
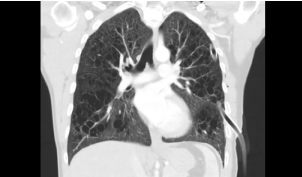
Figure 1: High-resolution computed tomography image in coronal plane
shows numerous bizarre shaped cysts of varying size in both lungs. Note that
only the lower zones were spared.
Pathological Findings
Macroscopically, a slight outward bulging with its greatest dimension being 1.2 cm was detected in pulmonary wedge resection material whose end was occluded with a stapler attracted attention. The surface of the slices cut from this area had a spongious structure containing very small cystic formations. All of the tissue was sampled. Microscopically, under small magnification marked congestion, and emphysematous changes, and slightly fibrotic alveolar septa adjacent to enlarged air ways in-between were more prominent (Figure 2). At only a few areas nodular formations enlarged, and extended into alveolar structures attracted our attention (Figure 3a). More attentive examination of these nodular structures revealed their bronchiolocentric characteristics (Figure 3b). Under greater magnification, these nodular formations were infiltrated with histiocytic cells with round-oval shaped, pale nuclei, and moderately enlarged acidophilic cytoplasm, and mixed type inflammatory cells relatively rich in lymphocytes (Figure 3c and 3d). More detailed examination revealed cells with similar formation which indistinctly infiltrated entire length of alveolar septa (Figure 4a and 4b). Patchy areas of fibrosis were detected. Immunohistochemical staining with CD1a, S-100 demonstrated marked uptake of dye by histiocytic cells (Figure 5). Not only scarce number of cells contained in nodules, but also, cells in the adjacent interstitial space were stained.
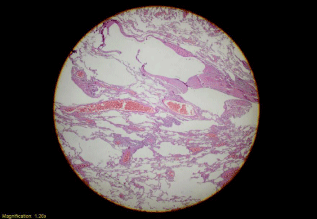
Figure 2: Low power magnification; marked congestion, emphysematous
changes and slightly fibrotic alveolar septa adjacent to enlarged air ways
(H&E x20).
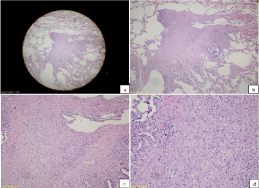
Figure 3: a) Nodular appearance, lower magnification (H&E x20), b) Note
the bronchiolocentric nature of nodular structures (H&E x40), c and d) Under
greater magnification, note that the nodular structures are composed of cells
rich in histocytic cells and rich in lymphocytes (H&E x100 and x200).
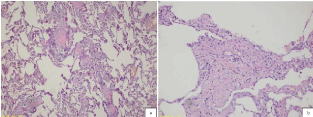
Figure 4: Note that a similar infiltration pattern is observed in the adjacent
alveolar septum (H&E x100 and x200).
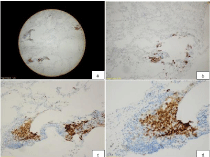
Figure 5: Immunohistochemical staining with CD1a in histiocytes (CD1a
x20, x40, x100 and x200).
Based on these findings, the final diagnosis was pulmonary LCH.
Discussion
Langerhans cells are a subtype of dendritic cells. Dendritic cells involve in the mononuclear fagocytic system, and they fulfill their functions by presenting antigen to T-lymphocytes. These cells are found in the skin, and mucosa, and normally they are scattered all along the airways. PLCH are the most important dendritic cell proliferation affecting the lungs. This condition is generally known as a form of interstitial pulmonary disease which is only confined to lungs, and it is accepted as a distinct entity from systemic LCH [3]. PLCH is generally an adult disease, and it is almost always seen in active smokers or quitters. Any risk factor is not known for the systemic form, and it is very frequently observed in small children [4].
PLCH has miscellaneous clinical presentations. Only 25% of the patients are asymptomatic, and they demonstrate characteristic signs in incidentally performed imaging modalities. In 15-25% of the cases the disease is complicated by spontaneous pneumothorax [5]. Our case has been followed up since 2014, and underwent open lung biopsy two years later because of development of pneumothorax. In the presence of typical radiological findings on HRCT scan (involvement of upper and middle pulmonary lobes, and relatively intact basal regions), and consistent clinical data (middle age, smoking history in a male patient) very frequently, the diagnosis of PLCH is successfully made [3]. Lung biopsy may not be required. However in cases with atypical radiological findings, and clinical presentation tissue biopsy may be required for definitive diagnosis. Open lung biopsy is more frequently regarded as a diagnostic tool for PLCH. These types of biopsies should be performed with the aid of CT (if possible), and biopsy materials should be obtained from multiple lobes. Biopsy material resected from only left inferosuperior segment was sent to us. If a marked nodular infiltrate can be macroscopically identified in an open pulmonary biopsy material obtained during wedge resection, the pathologist is lucky. Because the nodules are generally small in size, occasionally large nodules can be seen. In our case, macroscopically a prominent nodular formation was not seen, and very minute, cystic spongious pinhead-sized areas were observed. Microscopically, a conspicuous nodular structure was not found. Only, in addition to clinical follow-up, and imaging findings, very attentive microscopic evaluation allowed us to arrive at an accurate diagnosis.
In the early stage PLCH, cellular infiltration is marked, and typically forms loose cellular nodular structures scattered all over lung tissue, and spaces adjacent to small airways. At this stage, since bronchiolar wall, and neighbouring alveolar structures were exposed to destructive changes, small airways are subjected to progressive dilatation. With time, small airways are surrounded by hypocellular tissue rich in fibrotic tissue. Star-shaaped scar, and irregular parenchymal cystic lesions become conspicuous. Later Langerhans cells can not be detected in the lesion, and such lesions are defined as ‘‘burnt-out’’ PLCH.
Though rarely seen honeycomb changes are still observed in endstage disease. Since the patients with PLCH have a history of smoking, smoking-related changes in pulmonary parenchyma as respiratory bronchiolitis, Desquamative Interstitial Pneumonia (DIP), and/ or emphysema are seen. These conditions are considered in the histological differential diagnosis of PLCH. Detection of clusters of Langerhans cells with characteristic findings of morphology, and immunohistochemical staining allows exclusion of many disease states included in the differential diagnosis of PCHL. In the detection of PLCH, bronchiolocentric characteristic of the disease, enlargement of cystic airways, and presence of mixed inflammation are also very important markers. In the differential diagnosis spectrum of PLCH histiocytic/macrocytic lesions [DIP, respiratory bronchiolitis, Erdheim-Chester Disease (ECD), and hypersensitivity pneumonitis], and eosinophilic diseases (eosinophilic pneumonia) are taken into consideration. In all these diseases large groups and nodules of Langerhans cells are not seen. However in interstitium, and bronchiolar mucosa a few scattered Langerhans cells can be found. Besides, in all pulmonary diseases excepting PLCH histiocytes do not stain with CD1a, and langerin (CD207), they may only stained with S-100 dye. Lets analyse disease states considered in the differential diagnosis one by one. DIP is a completely diffuse process, rather than a nodular formation. Besides, cellular infiltration in DIP does not resemble to that seen in PLCH, and predominantly infiltration rich in macrophages is observed. In our case, we detected very minute indistinct nodular formations during very careful microscopic examination. This nodule was infiltrated with conspicuous lymphocytes, while in all other interstitial spaces faintly seen lymphocytes were observed. Apparent macrophage dominancy was not noted. In Eosinophilic Pneumonia (EP) presence of multiple number of eosinophils together with macrophages in airspace nodules is remarkable. Clusters of Langerhans cells are not seen. In ECD infiltration and/or nodules of histiocytes have/has a characteristic appearance. In ECD, histiocytes have a foamy cytoplasm, typical nuclear characteristics of Langerhans cells are not observed. Similar to PLCH, peribronchiolar space may contain histiocytes. However histiocytes demonstrate a lymphangitic distribution, and they are most frequently found in subpleural, interlobular septa, and perivascular areas. Advanced fibrotic stage of PLCH may be similar to the Usual manifestation of Interstitial Pneumonia (UIP). Contrary to UIP, PLCH has a predilection for subpleural space rather than centrilobular areas. Besides, star-shaped pattern of fibrotic nodules is a descriptive feature of PLCH, rather than UIP.
The most important treatment for PLCH is cessation of smoking. However still some cases progress or become stable despite continuation of smoking. Nowadays, any predictive biomarker which can fully foresee progression of PLCH is not available. In some studies beneficial effects of corticosteroids, and chemotherapeutic agents in the management of severe or progressive disease have been reported. Cladribine (2-chlorodeoxyadenosine ), an agent cytotoxic to lymphocytes and monocytes, has been used in LCH and recently has also been tried for a few patients with PLCH. However, these medications seem to be of limited value additional data are also needed to assess the efficacy of these agents [6]. Lung transplant might be considered for patients with advanced, progressive disease. But, PLCH can recur after lung transplantation.
It is debatable whether PLCH is a reactive or a neoplasic condition. Since in most of the patients it is associated with smoking, it was thought to be a non-neoplasic disease. However in systemic LCH, any provocative agent has not been clearly defined. The presence of clonality in systemic LCH was demonstrated in a study by Willman et al. performed on 10 patients in the year 1994 [7]. Further studies have also provided additional support for the clonality of LCH [8,9]. More recently, in two studies the presence of BRAF V600E expression has been described which will substantiate the assertion that systemic LCH might be a type of myeloid neoplasm [10,11].
Two different studies performed by Yousem et al. have also supported the presence of clonality in PLCH [12,13]. Later on Roden et al. defined BRAF V600E expression in 28% of PLCH, and 35 % of systemic LCH patients [14].
References
- Shen J, Feng S. Bone Langerhans cell histiocytosis with pulmonary involvement in an adult non-smoker: A case report and brief review of the literature. Molecular and Clinical Oncology. 2017; 6: 67-70.
- Dejima H, Morita S, Takahashi Y, Matsutani N, Iinuma H, Kondo F, et al. A case of invasive Langerhans cell histiocytosis localizing only in the lung and diagnosed as pneumothorax in an adolescent female. International journal of clinical and experimental pathology. 2015; 8: 3354-3357.
- Roden AC, Yi ES. Pulmonary Langerhans Cell Histiocytosis: An Update From the Pathologists’ Perspective. Archives of pathology & laboratory medicine. 2016; 140: 230-240.
- Mason RH, Foley NM, Branley HM, Adamali HI, Hetzel M, Maher TM, et al. Pulmonary Langerhans cell histiocytosis (PLCH): a new UK register. Thorax. 2014; 69: 766-767.
- Mendez JL, Nadrous HF, Vasallo R, Decker P, Ryu JH. Pneumothorax in Pulmonary Langerhans’ Cell Histiocytosis. CHEST Journal 2003; 124: 108S.
- Grobost V, Khouatra C, Lazor R, Cordier JF, Cottin V. Effectiveness of cladribine therapy in patients with pulmonary Langerhans cell histiocytosis. Orphanet journal of rare diseases. 2014; 9: 191.
- Willman CL, Busque L, Griffith BB, Favara BE, McClain KL, Duncan MH, et al. Langerhans’-cell histiocytosis (histiocytosis X)-a clonal proliferative disease. New England Journal of Medicine. 1994; 331: 154-160.
- Tatsuno M, Shioda Y, Iwafuchi H, Yamazaki S, Iijima K, Takahashi C, et al. BRAF V600 mutations in Langerhans cell histiocytosis with a simple and unique assay. Diagn Pathol. 2016; 11: 39.
- Gong L, Zhang W, Li Y, Liu XY, Yao L, Han XJ, et al. Clonal status and clinicopathological features of Langerhans cell histiocytosis. Journal of International Medical Research. 2010; 38: 1099-1105.
- Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010: 116: 1919-1923.
- Sahm F, Capper D, Preusser M, Meyer J, Stenzinger A, Lasitschka F, et al. BRAFV600E mutant protein is expressed in cells of variable maturation in Langerhans cell histiocytosis. Blood. 2012; 120: e28-e34.
- Yousem SA, Colby TV, Chen YY, Chen WG, Weiss LM. Pulmonary Langerhans’ cell histiocytosis: molecular analysis of clonality. The American journal of surgical pathology. 2001; 25: 630-636.
- Yousem SA, Dacic S, Nikiforov YE, Nikiforova M. Pulmonary Langerhans cell histiocytosis: profiling of multifocal tumors using next-generation sequencing identifies concordant occurrence of BRAF V600E mutations. CHEST Journal. 2013; 143: 1679-1684.
- Roden AC, Hu X, Kip S, Parrilla Castellar ER, Rumilla KM, Vrana JA, et al. BRAF V600E expression in Langerhans cell histiocytosis: clinical and immunohistochemical study on 25 pulmonary and 54 extrapulmonary cases. The American journal of surgical pathology. 2014; 38: 548-551.